
文献解读
客户案例Circulation | 氘代丝氨酸代谢流如何证实一碳代谢酶不靠代谢也致病,肺动脉高压全新靶点
肺动脉高压(PH)属于恶性心血管疾病,核心病理特征为持续性肺血管结构重塑、肺循环压力升高,最终诱发不可逆右心衰竭。当前临床靶向药物多仅能发挥单纯肺血管扩张作用,难以从根源阻断血管重构进程,患者远期预后较差。因此,挖掘调控肺血管内皮稳态的全新分子通路、开发针对性干预靶点成为PH领域亟待突破的关键方向。
2026年6月,山东大学研究团队在Circulation上在线发表了题为”Endothelial SHMT2 Drives Pulmonary Vascular Remodeling Through Noncanonical Pathway in Pulmonary Hypertension”的研究文章,采用”临床蛋白质组学发现-体内功能验证-非代谢机制解析-虚拟筛选药物-疗效验证“的全链条研究思路,阐明了SHMT2(丝氨酸羟甲基转移酶2)通过其非经典代谢功能——作为BRISC去泛素化酶复合物组分,去除RhoB蛋白K181位点的K63连接泛素链,抑制RhoB溶酶体降解,从而促进内皮屏障功能障碍和血管重构,为理解PH发病机制和干预靶点提供新视角。(麦特绘谱提供代谢流[2,3,3-D3]- Serine检测服务)
研究设计
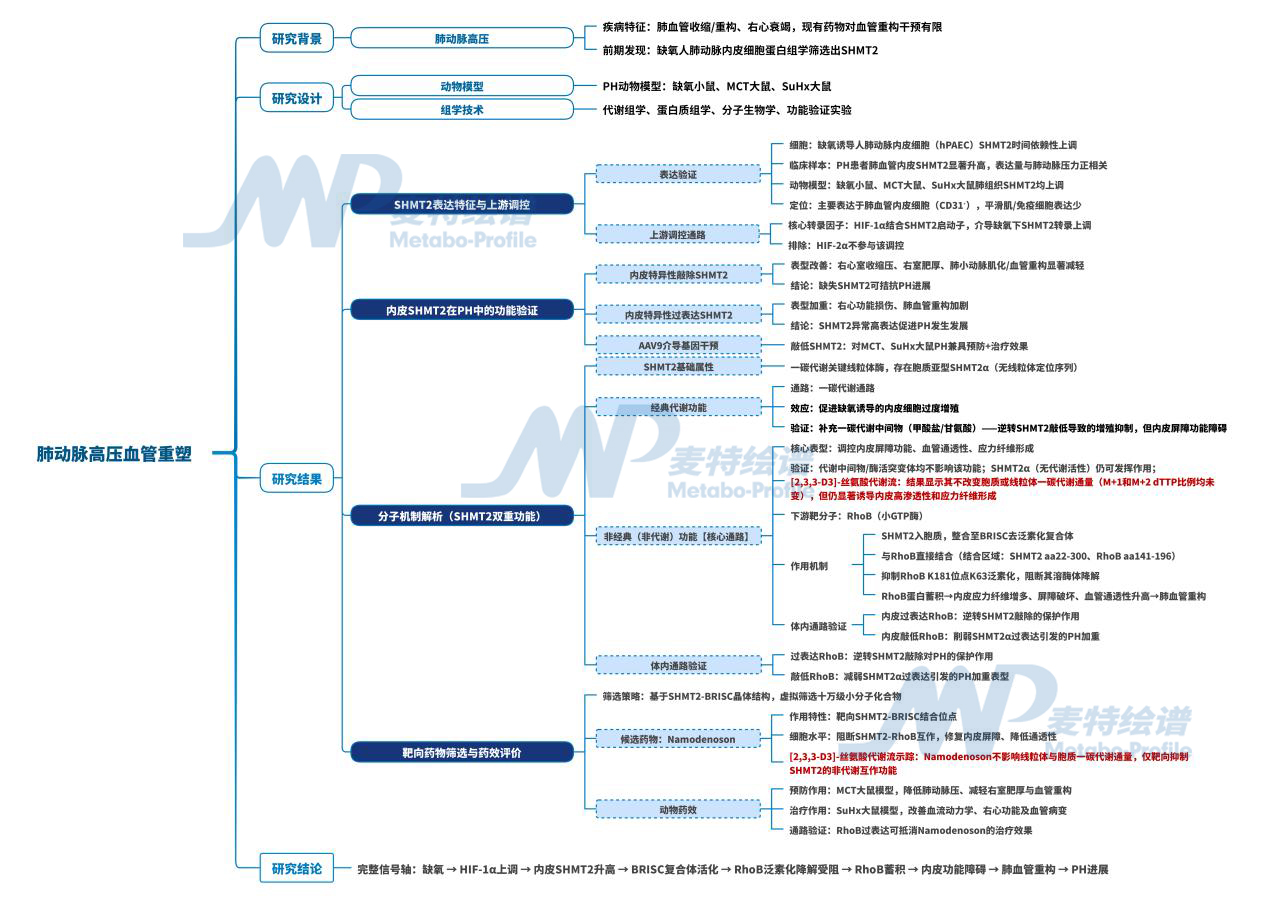
图1. 技术路线
研究结果
1. 蛋白质组学发现关键线索:SHMT2是与PH相关的致病蛋白
为了寻找与PH相关的关键蛋白,首先对缺氧处理的人肺动脉内皮细胞(hPAEC)进行了蛋白质组学分析,鉴定出1287个差异表达蛋白。通过三重筛选标准(缺氧显著上调、肺/心脏高表达、参与代谢调控),锁定SHMT2作为候选靶点。
为验证其临床相关性,对PH患者肺组织进行免疫荧光染色,发现SHMT2主要表达于内皮细胞,且其表达水平与平均肺动脉压呈显著正相关。在三种PH模型(缺氧小鼠、MCT大鼠、SuHx大鼠)的肺组织中,SHMT2蛋白表达均显著上调。体外实验进一步证实,缺氧处理hPAEC可时间依赖性诱导SHMT2表达升高。这些结果表明,内皮SHMT2是PH的潜在致病因子。
针对上游调控通路的实验显示,缺氧可同步升高HIF‑1α与HIF‑2α,但只有敲低HIF‑1α能完全阻断缺氧带来的SHMT2上调,ChIP‑qPCR实验进一步直接证实,缺氧环境下HIF‑1α可特异性结合SHMT2基因启动子区域,直接启动其转录过程。
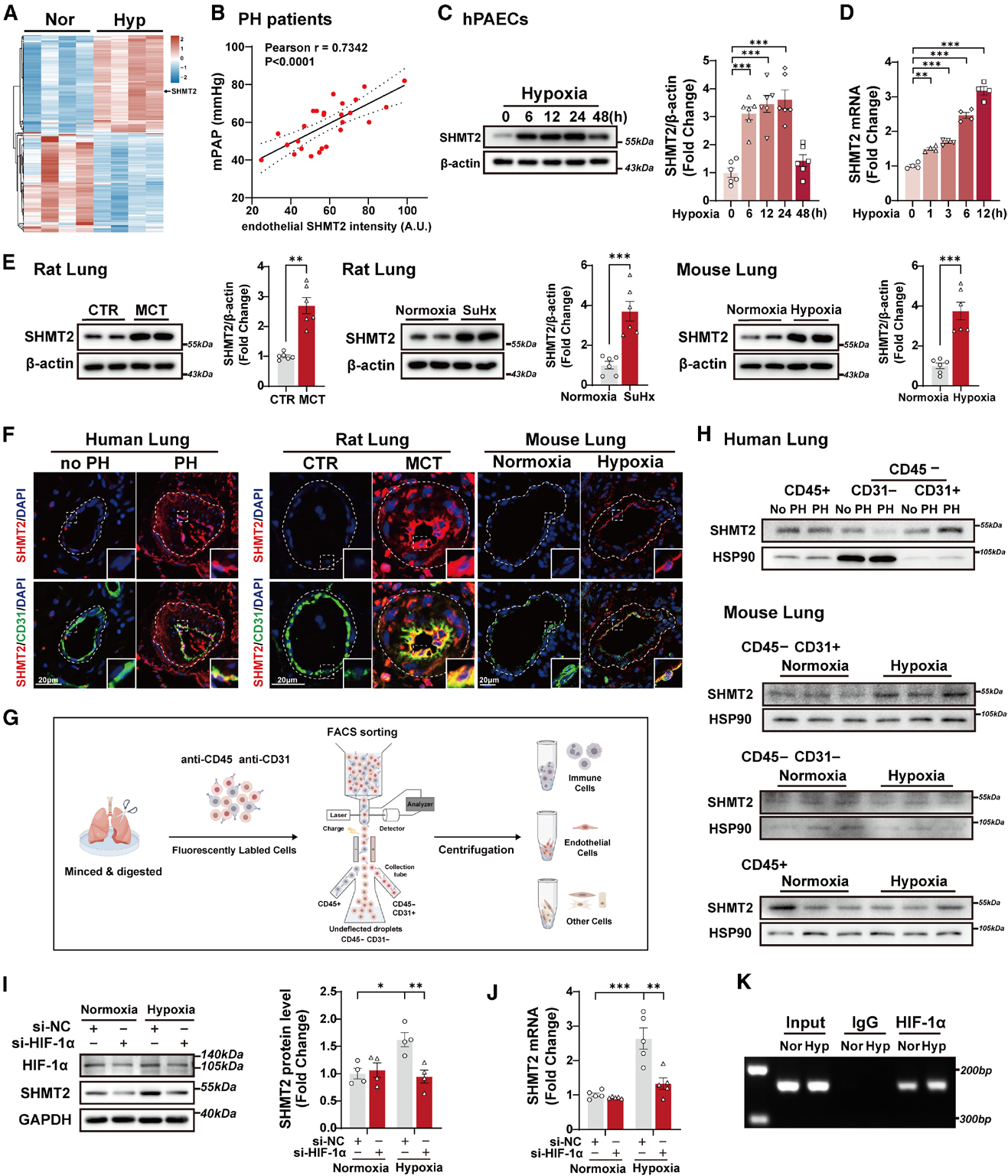
图2. 内皮细胞SHMT2在多种PH模型中上调
2. 内皮特异性敲除SHMT2缓解PH,过表达加重PH
为了明确SHMT2在PH中的功能,研究者构建了内皮细胞特异性Shmt2敲除小鼠(Shmt2ECKO)和AAV9-ICAM2介导的内皮特异性过表达模型。缺失保护效应方面:Shmt2ECKO小鼠常氧下无表型,缺氧后右室收缩压(RVSP)、肺动脉肌化等指标均显著低于对照。过表达致病效应方面:AAV9-ICAM2-Shmt2小鼠缺氧后RVSP、右心室肥厚、肺动脉肌化显著加重。
为进一步验证治疗潜力,研究者在大鼠模型中开展基因治疗(注射AAV9-Shmt2-shRNA):MCT大鼠(造模前2周给药)mPAP、RVSP、壁厚度、肌化比例、RV肥厚均显著改善;SuHx大鼠(PH建立后给药)同样显著缓解血流动力学和病理指标。这些结果证实内皮SHMT2是PH发生发展的必需驱动因子,SHMT2沉默对PH兼具预防和治疗价值。
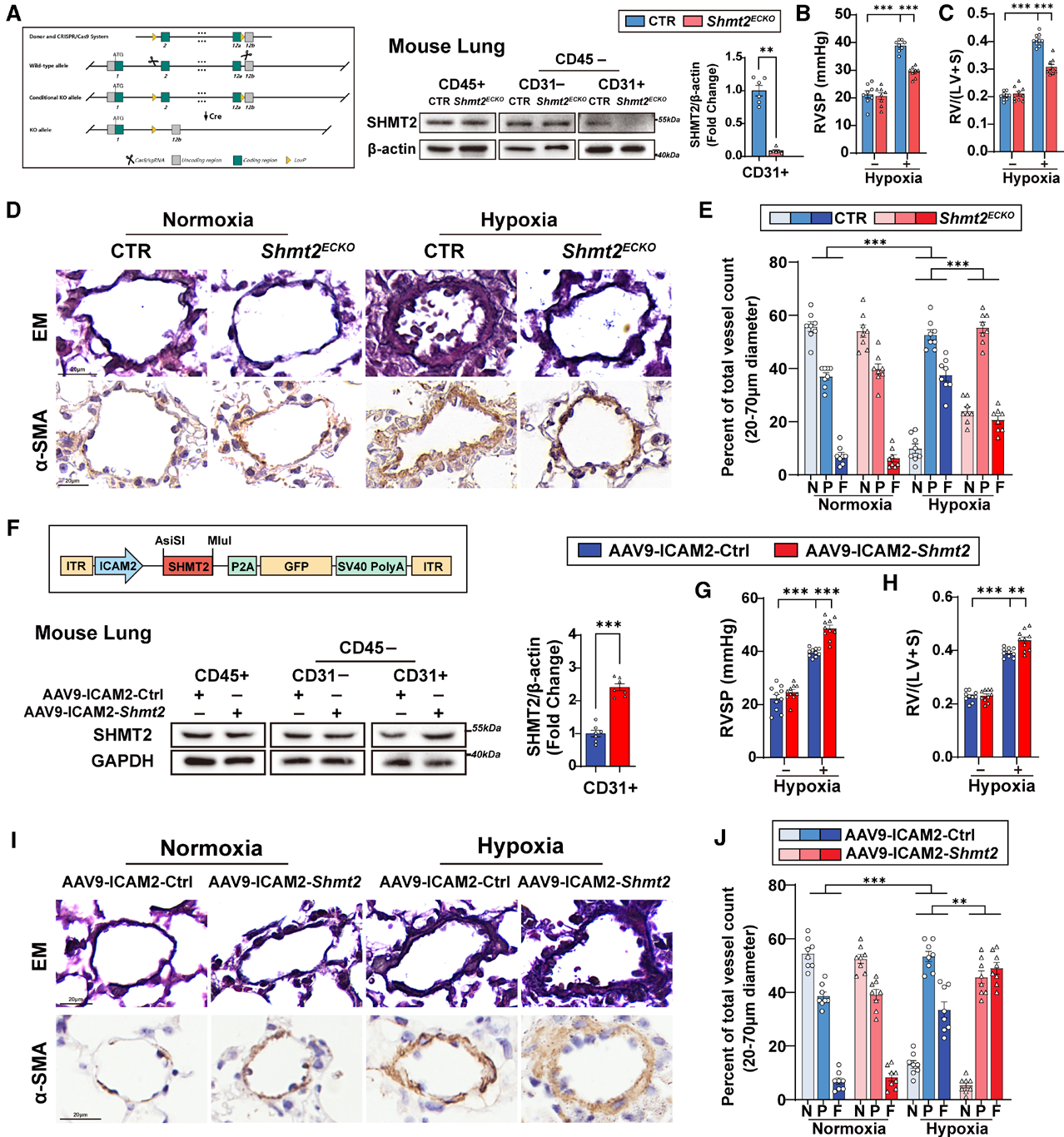
图3. 内皮细胞特异性缺失SHMT2可改善低氧诱导的小鼠PH,而SHMT2过表达则加重PH
3. 非经典功能揭秘:SHMT2通过非代谢机制调控内皮屏障
为了解析SHMT2调控内皮功能的分子机制,对si-SHMT2的蛋白质组学结果进行分析,发现差异蛋白主要集中在应力纤维组装、内皮屏障稳态、上皮间质转化相关通路,且差异蛋白多分布于细胞质与细胞膜,线粒体占比仅6.7%,提示SHMT2的功能可能超越经典代谢范畴。
为进一步验证SHMT2经典代谢与非代谢功能,开展了一系列功能实验:
补充一碳代谢中间体(甲酸盐+甘氨酸)可挽救SHMT2敲除导致的增殖缺陷,但无法挽救内皮屏障功能障碍;
SHMT2酶活突变体K95Q/K280Q(这两个位点是SHMT2酶活性中心的关键残基,突变后SHMT2失去催化丝氨酸→甘氨酸的能力)过表达仍可逆转SHMT2敲除的屏障保护效应;
胞质异构体SHMT2α(无线粒体靶向序列,无法进入线粒体)过表达:[2,3,3-D3]-丝氨酸代谢流结果显示其不改变胞质或线粒体一碳代谢通量(M+1和M+2 dTTP比例均未变),但仍显著诱导内皮高渗透性和应力纤维形成。
体内验证显示,AAV9-ICAM2-Shmt2α过表达加重缺氧诱导的RVSP、RV肥厚和血管重塑,TEM显示内皮紧密连接破坏。这些结果表明SHMT2促进缺氧诱导的内皮屏障功能障碍主要依赖非经典功能,即非其一碳代谢酶活性。
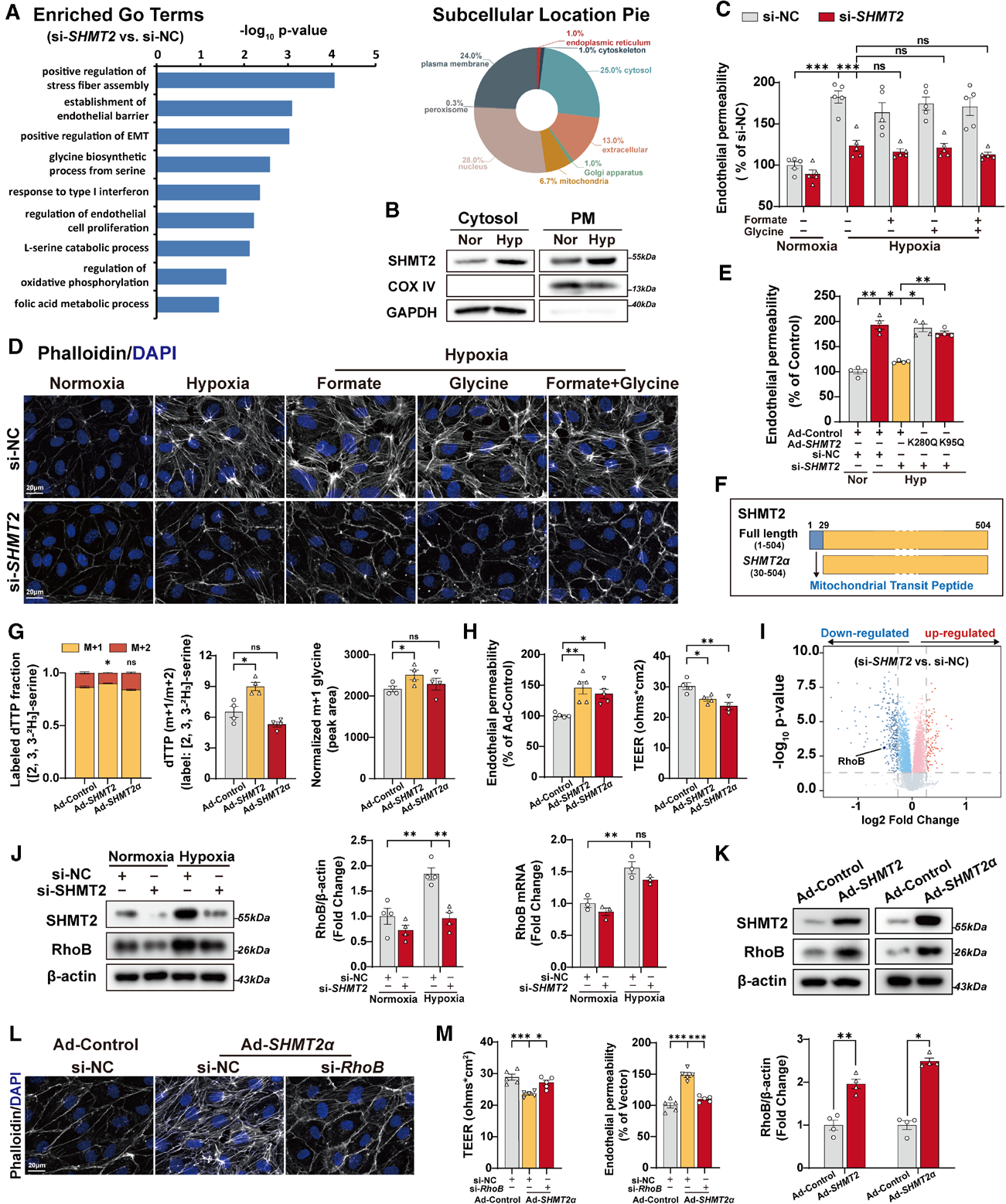
图4. SHMT2通过非经典代谢功能,介导缺氧诱导的hPAECs内皮屏障的破坏
4. RhoB是SHMT2非经典功能的关键介导分子
为了找出SHMT2非经典功能的下游效应分子,对si-SHMT2蛋白质组学结果进行筛选发现:RhoB——其在SHMT2敲低后蛋白丰度显著下调,但mRNA水平无变化,说明SHMT2在蛋白降解环节调控RhoB。
细胞双向验证实验显示,SHMT2与胞质SHMT2α过表达均可上调RhoB蛋白;细胞回补实验证实,敲低RhoB能够完全消除SHMT2α过表达引发的应力纤维增多与内皮高渗漏。体内通路回补实验分为两组,在内皮Shmt2敲除小鼠中特异性过表达RhoB,原本敲除带来的肺动脉高压保护效应被完全逆转,小鼠右室压力、右心肥厚、血管肌化与肺内皮渗漏再次加重,电镜可见内皮连接破损;反之,在SHMT2α内皮过表达小鼠中特异性敲低RhoB,肺动脉高压表型显著缓解,伊文思蓝渗漏减少,内皮紧密连接结构恢复完整,证实SHMT2通过调控RhoB驱动肺血管重构。
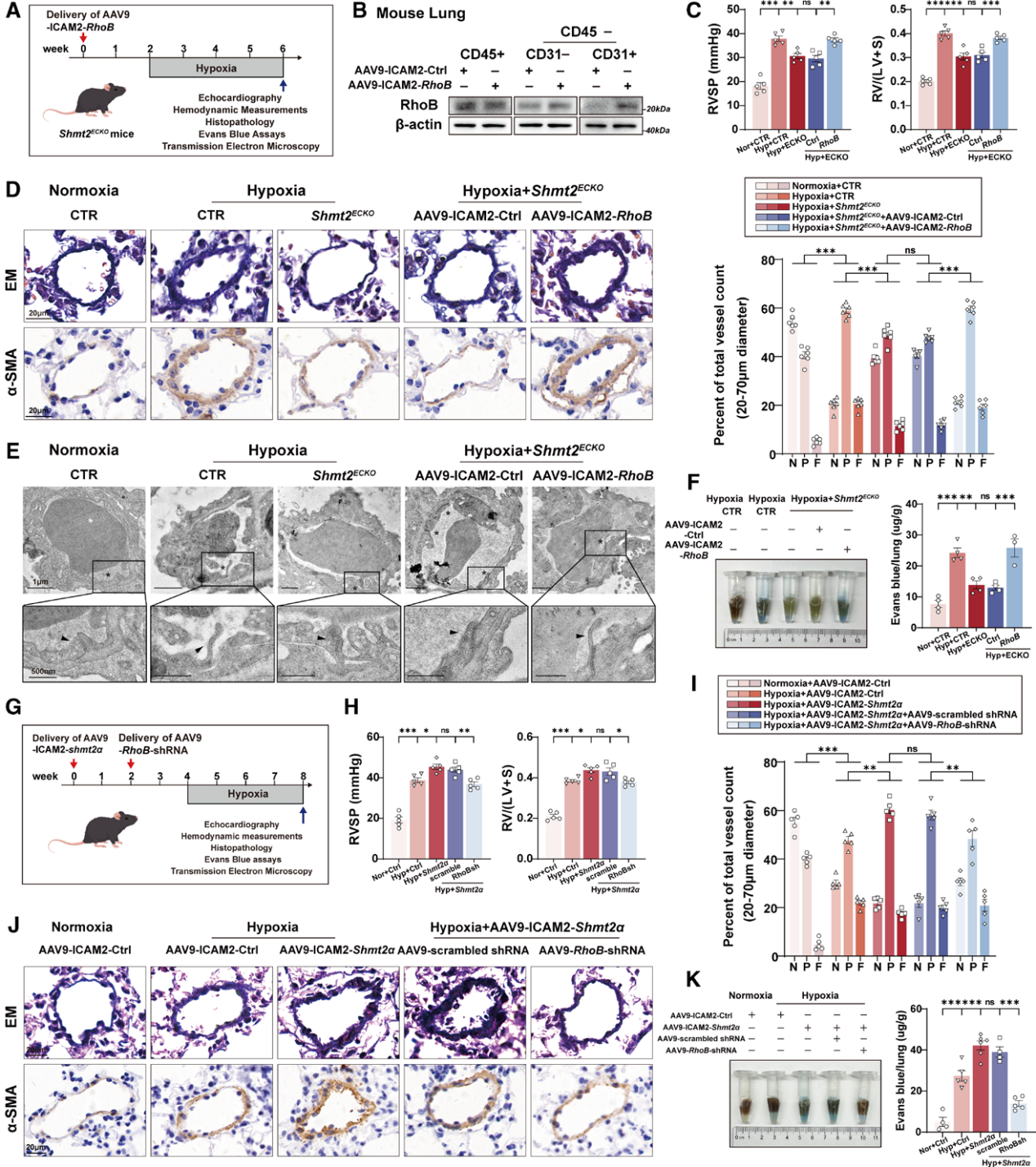
图5. RhoB在体内介导SHMT2相关的内皮屏障功能障碍和肺动脉高压
5. SHMT2-BRISC复合物去除RhoB的K63泛素化
在确认RhoB是SHMT2下游关键效应分子,且SHMT2通过非代谢功能调控RhoB蛋白水平后,研究进一步深入探究了SHMT2究竟如何调控RhoB蛋白稳定性的分子机制。
降解途径鉴定:SHMT2敲除后RhoB mRNA水平不变而蛋白水平下降,提示存在转录后调控。放线菌酮(CHX)追踪实验证实SHMT2/SHMT2α显著延长RhoB蛋白半衰期。使用多种降解途径抑制剂发现,只有溶酶体抑制剂(氯喹、NH4Cl)能阻断SHMT2敲除诱导的RhoB下降,而蛋白酶体抑制剂均无效,表明SHMT2通过抑制溶酶体途径降解来稳定RhoB。
蛋白互作及结合位点定位:共免疫沉淀(Co-IP)证实SHMT2与RhoB直接结合,缺氧增强二者相互作用。共聚焦显微镜显示二者在hPAEC中存在明显共定位。通过截短突变体实验精确定位:SHMT2的N端区域(aa 22-300)和RhoB的C端区域(aa 141-196)介导二者结合。
BRISC复合物介导去泛素化机制:SHMT2为BRISC去泛素化复合物组分,BRCC36为该复合物催化亚基;敲低BRCC36可阻断SHMT2α上调RhoB的作用。泛素化检测显示:SHMT2/SHMT2α负向调控RhoB泛素化水平;机制分型证实,该复合物特异性清除RhoB K63型多聚泛素链,不作用于K48泛素链,契合K63泛素链介导底物溶酶体降解的经典机制。
泛素化关键靶位点验证:针对RhoB结合区段(141-196aa)4个赖氨酸位点定点突变,结果显示K181位点为核心功能位点:RhoB-K181R突变体本底K63泛素化水平极低,且SHMT2无法调控该突变体泛素化水平,证实K181是BRISC复合物去除RhoB K63泛素化的核心位点。
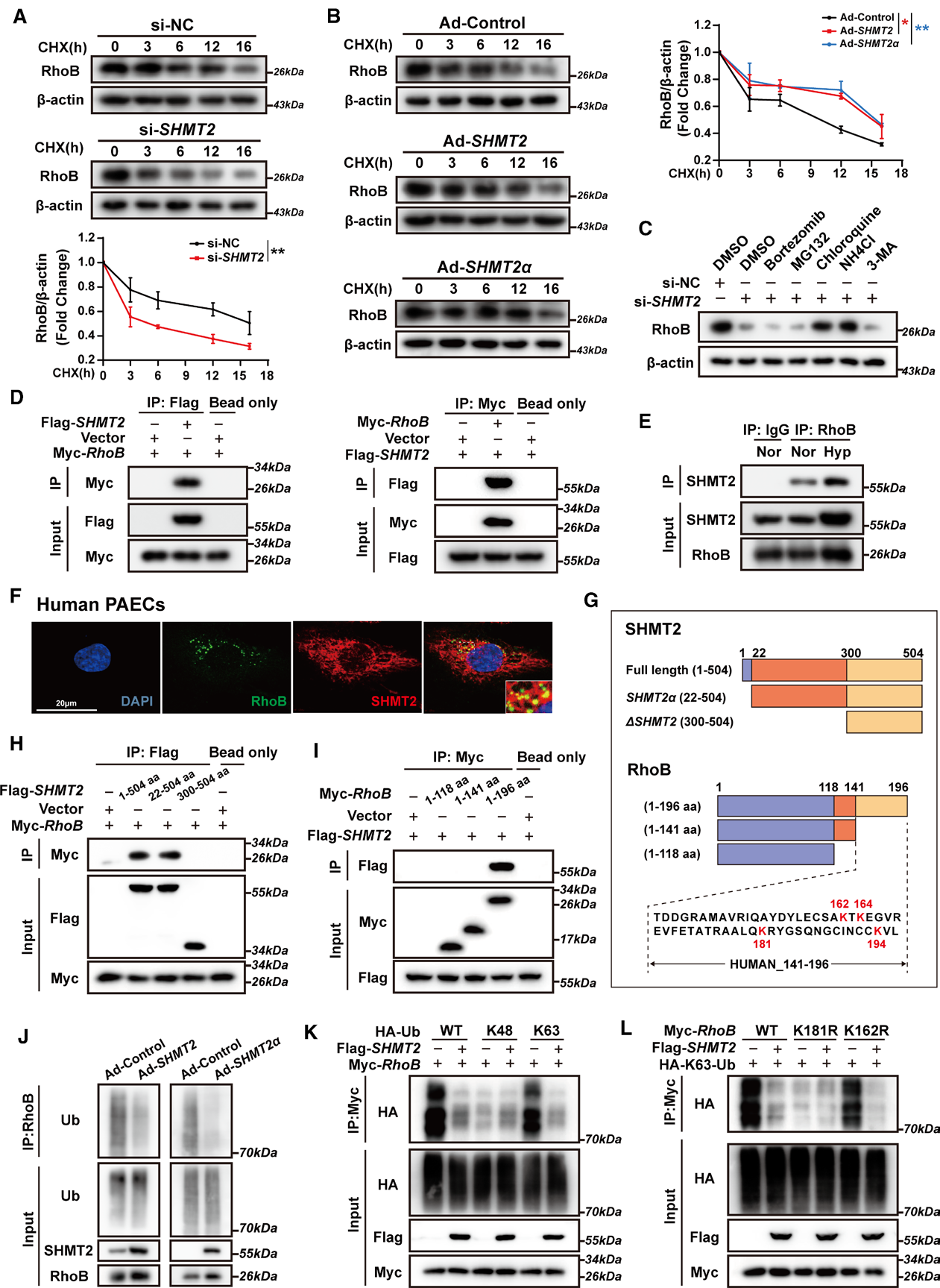
图6. RhoB的K181残基是SHMT2抑制K63连接的RhoB泛素化的关键
6. 虚拟筛选与药物验证:Namodenoson靶向SHMT2-RhoB通路
研究依托SHMT2-BRISC复合体晶体结构开展十万级小分子虚拟筛选,初筛得到200个候选化合物,结合临床应用前景缩小至27个,最终筛选出Namodenoson。体外细胞分子实验显示,Namodenoson可阻断 SHMT2 与RhoB的相互作用,恢复RhoB的K63泛素化修饰,加速RhoB降解;[2,3,3‑D3]-丝氨酸代谢流示踪结果证实,Namodenoson不影响线粒体与胞质一碳代谢通量,仅靶向抑制SHMT2的非代谢互作功能。
在MCT大鼠预防模型和SuHx大鼠治疗模型中,Namodenoson都展现出改善PH指标的治疗效果。靶点特异性验证表明,MCT联合AAV9-ICAM2-Shmt2α大鼠中Namodenoson仍显著改善血管重塑,但内皮RhoB过表达(AAV9-ICAM2-RhoB)显著削弱其保护效应。这些结果表明Namodenoson通过靶向SHMT2-RhoB非经典通路,发挥预防和治疗PH的双重作用,具有良好的临床转化潜力。
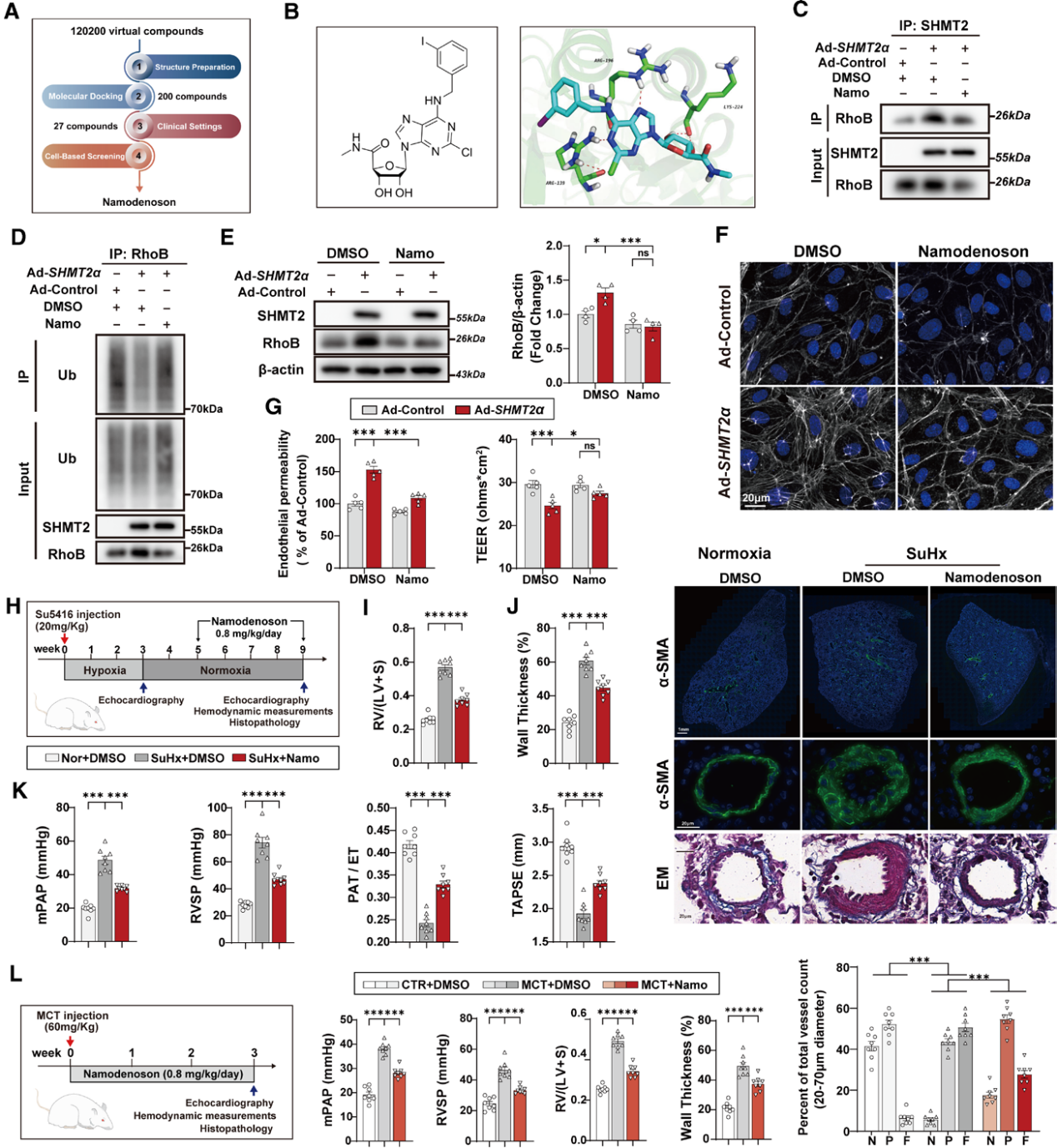
图7. Namodenoson为SHMT2非经典功能的小分子抑制剂及其在PH中的治疗潜力
研究结论
缺氧诱导内皮SHMT2上调,SHMT2作为BRISC去泛素化酶复合物的组分,通过结合RhoB蛋白K181位点,去除其K63连接多聚泛素链,抑制RhoB溶酶体降解,导致RhoB蛋白积累,进而促进应力纤维形成和内皮屏障功能障碍,最终驱动肺血管重构和肺动脉高压。
参考文献
Endothelial SHMT2 Drives Pulmonary Vascular Remodeling Through Noncanonical Pathway in Pulmonary Hypertension. Circulation. 2026
扫描二维码阅读原文

绘谱帮你测
本研究中通过麦特绘谱提供的[2,3,3‑D3]-丝氨酸代谢流技术验证了SHMT2并非依靠经典代谢途径介导内皮屏障损伤,而是依赖其胞质亚型介导的BRISC复合体- RhoB非经典通路发挥致病效应,也为后续靶向Namodenoson阻断SHMT2非代谢功能、不干扰机体正常一碳代谢的治疗思路提供了关键代谢证据支撑。
麦特绘谱开创性地搭建了医学领域高端代谢组学技术平台,覆盖了非靶向-全定量-代谢流等全方位的高端医学代谢组解决方案,同时全面布局微生物组学、转录组学和蛋白质组学等多组学技术服务,已成为全球多组学研究者的优选合作伙伴。麦特绘谱已为数百家三甲医院、科研院所和企业提供多组学一站式整体解决方案,协助客户与合作伙伴发表SCI文章700+篇,累计影响因子7000+,平均IF>10,涵盖Cell, Science, Nature, Cancer Cell, Signal Trans-duction and Targeted Therapy, Nature Biotechnology, Cell Metabolism等顶级期刊。
