
文献解读
Cancer Research | 抑制KRAS信号导致胰腺癌产生可靶向的依赖脂噬的脂肪酸氧化的代谢依赖性
胰腺导管腺癌(PDAC)最显著的分子特征之一是高频的致癌KRAS突变,其持续激活RAF-MEK-ERK通路驱动肿瘤进展。近年来,针对KRAS及其下游MEK、ERK信号的多种抑制剂已进入临床开发阶段,但胰腺癌对这些靶向治疗普遍存在内在的代谢适应性和耐药性,导致单一靶点治疗效果有限。代谢重编程是肿瘤耐药的关键机制之一,然而,此前研究很少系统性地探究KRAS信号抑制引起的全面代谢通路重塑,尤其是脂质代谢的重塑机制。
近日,美国俄克拉荷马大学健康科学中心Pankaj K. Singh等研究团队在期刊Cancer Research(IF=16.6)发表题为“KRAS Signaling Inhibition Induces a Targetable Metabolic Dependency on Lipophagy-Dependent Fatty Acid Oxidation in Pancreatic Cancer”,该研究综合运用代谢组学、脂质组学和同位素示踪代谢流技术,系统解析了胰腺癌在ERK抑制后的代谢重编程特征,首次揭示了转录因子TFEB介导的脂噬-脂肪酸氧化通路是胰腺癌抵抗KRAS靶向治疗的核心代谢逃逸机制,并证实了联合抑制KRAS/MEK/ERK信号与脂肪酸氧化可产生协同抗肿瘤效应,为胰腺癌的精准治疗提供了全新的代谢靶点策略。

技术路线图

研究结果
1. ERK抑制增强胰腺癌细胞脂肪酸氧化
非靶向代谢组结果表明,在多种PDAC细胞系中,对照组和ERK抑制剂(ERKi)组的代谢特征存在明显差异,具体体现在:ERKi组的酰基肉碱类代谢物含量显著增加,而糖酵解相关代谢物显著减少,谷氨酰胺和谷氨酸水平下降。C13全标棕榈酸代谢流实验进一步验证,ERKi组中乙酰辅酶A和TCA循环中间产物的M+2标记占比显著增加,说明ERK抑制处理可以增强胰腺癌细胞脂肪酸氧化。
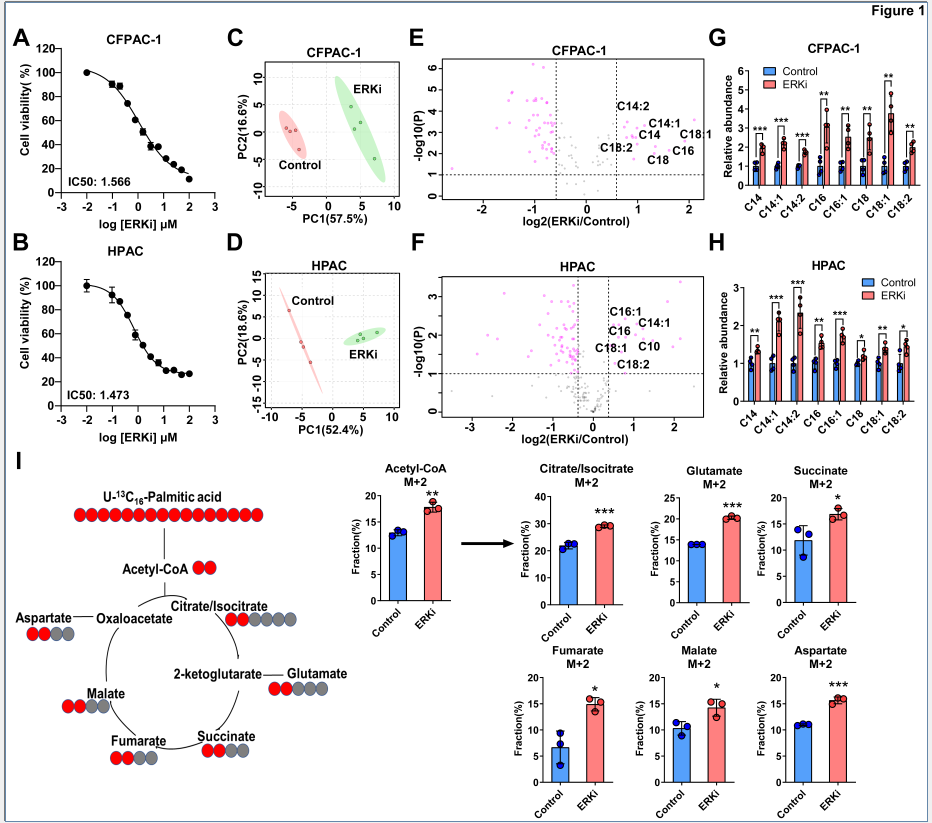
图1. ERK抑制增强胰腺癌细胞系中的脂肪酸氧化
2. ERK抑制加速脂质周转以供给脂肪酸氧化
脂质组学结果表明:(1)在细胞内,ERK抑制处理导致甘油三酯(TG)含量显著降低,溶血磷脂酰胆碱(LPC)含量大幅升高,游离脂肪酸(FA)含量也略有上升;(2)在细胞外,ERKi处理组与对照组的培养基中FA、TG水平以及FA摄取率上均无显著差异,证实ERK抑制后脂肪酸氧化的增强与外源脂质摄取无关。D31-棕榈酸脉冲追踪实验表明,ERKi处理组标记TG的分解速度显著加快,同时标记酰基肉碱的强度和比例均显著增加。使用脂肪酸氧化抑制剂可以显著降低ERKi处理细胞的存活率,这表明脂质周转驱动的脂肪酸氧化是ERK抑制后胰腺癌细胞的生存依赖通路。
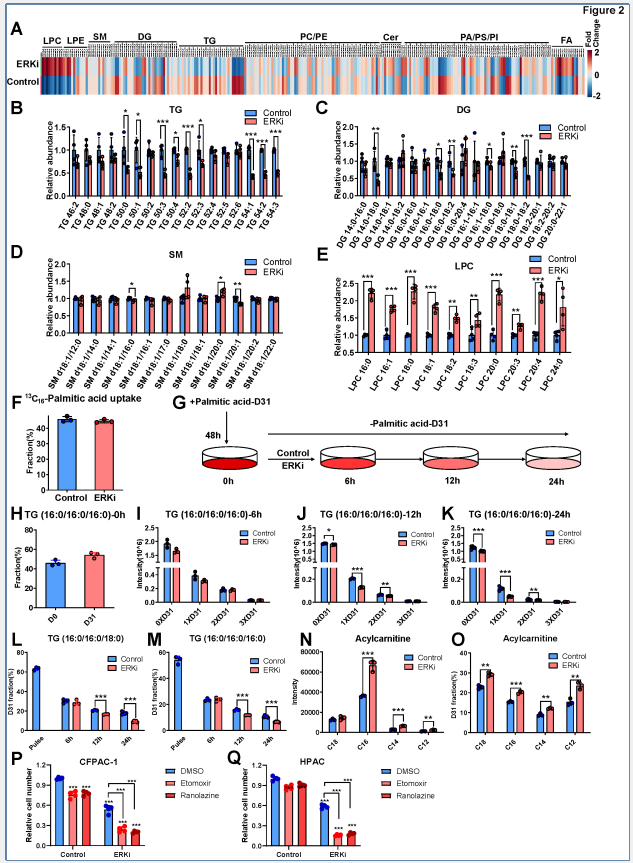
图2. ERK抑制加速脂质转换用于为脂肪酸氧化提供底物
3. ERK抑制上调脂噬,促进脂肪酸氧化
脉冲追踪实验表明:脂解抑制剂对ERKi诱导的脂质周转和脂肪酸氧化无显著影响;而脂噬抑制剂完全逆转了ERKi对脂质周转和脂肪酸氧化的促进作用。siRNA敲低实验证实:敲低LIPA(脂噬关键酶)可有效阻断ERKi诱导的脂质周转加速和脂肪酸氧化增加,而敲低PNPLA2(脂解关键酶)则无此效果。免疫荧光实验也显示,ERKi处理后总脂滴数量减少,而与溶酶体共定位的脂滴(即脂噬现象)显著增加。
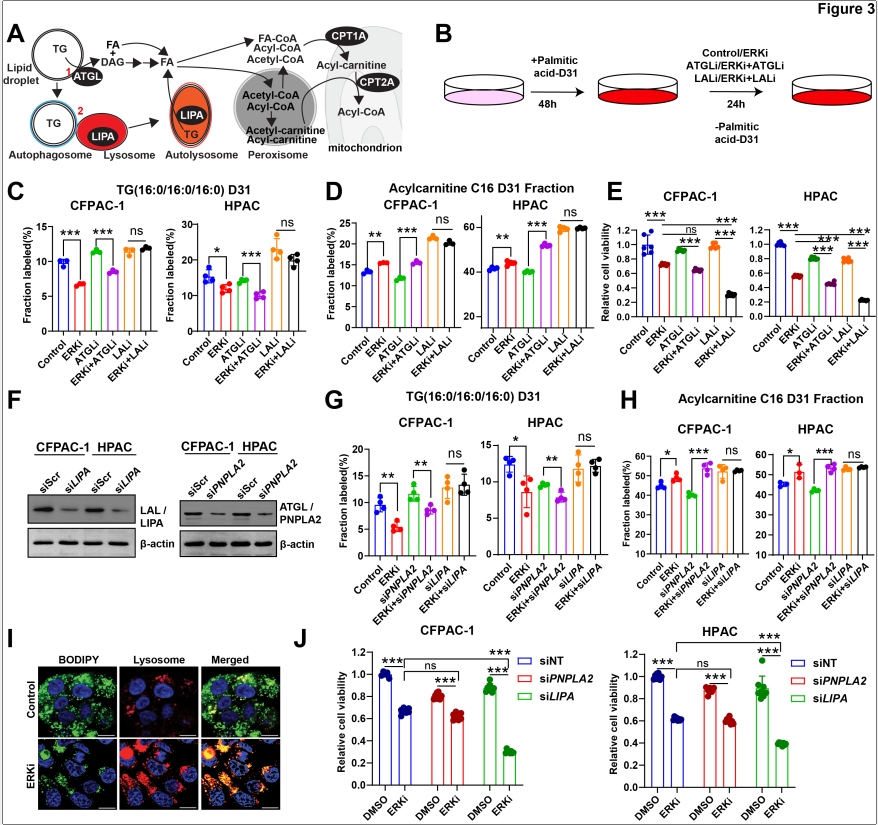
图3. ERK抑制上调脂噬
4. TFEB调控ERK抑制下的脂肪酸氧化和脂噬
qPCR定量表明,ERKi处理后脂肪酸氧化基因和脂噬基因的表达均显著上调。通过分析之前研究PDAC细胞系的RNA-seq数据发现,ERK 抑制通过转录上调参与代谢途径的基因来促进脂滴吞噬作用,从而为脂肪酸氧化提供能量。在潜在调控因子筛选中,研究者发现ERK抑制显著促进了转录因子TFEB的核转位。ChIP-PCR实验证实,ERK抑制后TFEB与CLEAR元件的结合显著增强。TFEB敲低实验证实,shRNA介导的TFEB沉默完全消除了ERKi诱导的脂噬和脂肪酸氧化基因上调,并使细胞对ERK抑制更为敏感。
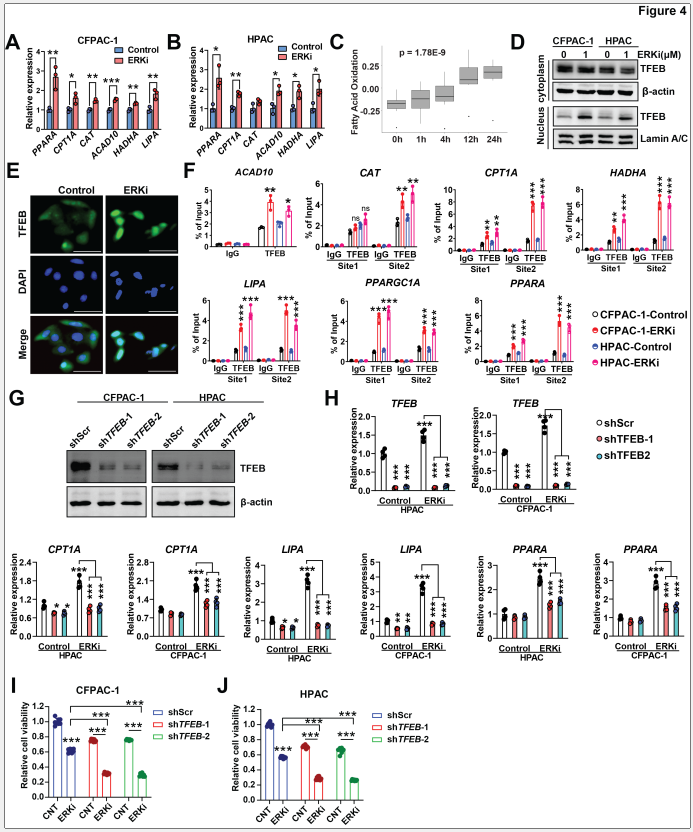
图4. ERK抑制通过TFEB上调脂噬和脂肪酸氧化的转录特征
5. 脂肪酸氧化抑制增强MEK/ERK抑制剂疗效
ERK抑制剂与脂肪酸氧化抑制剂联合使用时可显著增强细胞生长抑制效应,额外降低20-40%的细胞活力。联合指数(CI)分析显示两者具有强协同效应。通过shRNA敲低脂肪酸氧化限速酶CPT1A,证实遗传学阻断脂肪酸氧化同样可显著增加PDAC细胞对ERKi和MEKi的敏感。
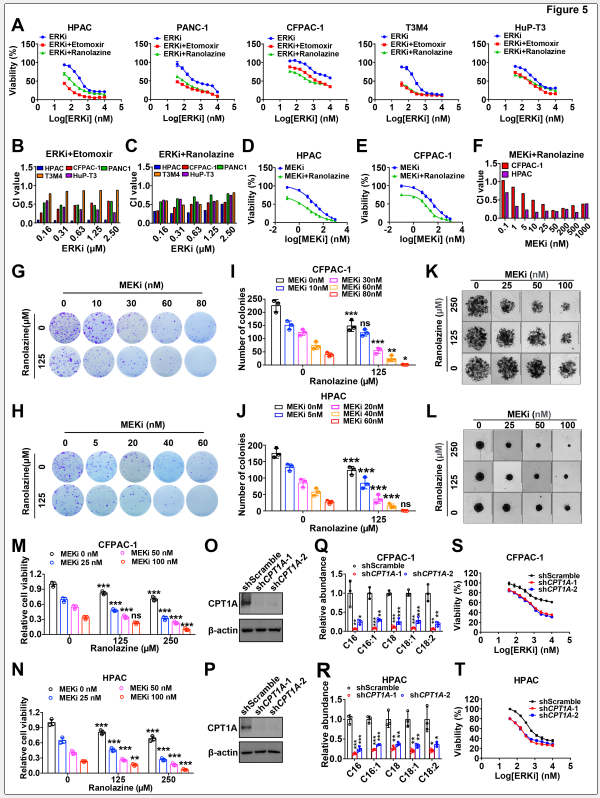
图5. 脂肪酸氧化抑制使胰腺癌细胞对MEK/ERK抑制剂敏感化
6. 体内验证:联合治疗显著抑制胰腺肿瘤生长
MEK抑制剂与脂肪酸氧化抑制剂联合治疗显著抑制了小鼠肿瘤生长,免疫组化分析显示Ki67阳性细胞显著减少,cleaved Caspase 3(凋亡标志物)染色显著增加。此外,血液生化指标显示联合治疗未产生明显毒性。
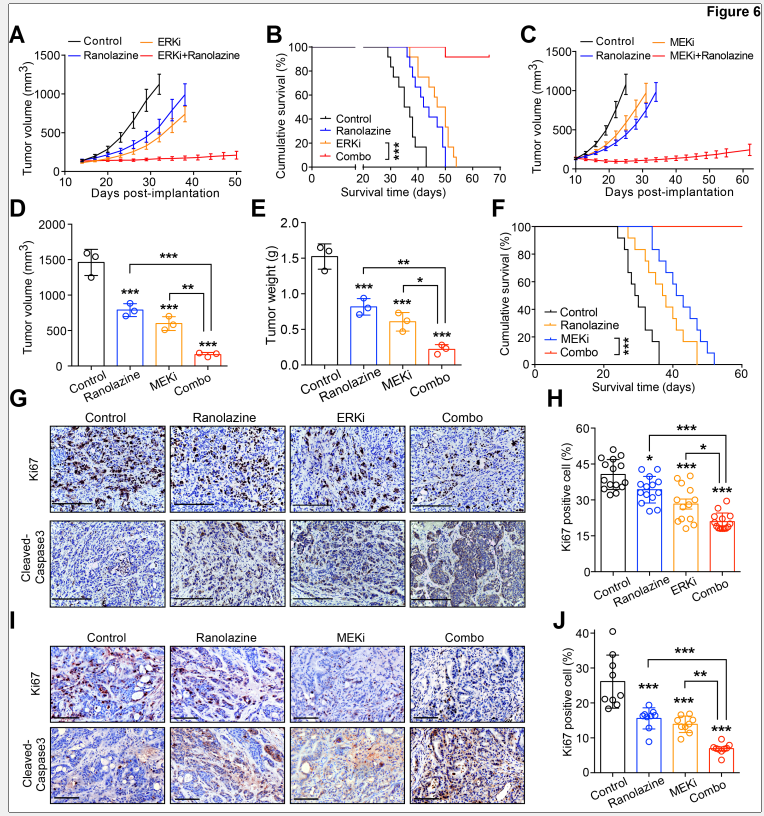
图6. 脂肪酸氧化抑制与MEK/ERK抑制剂联合使用在体内抑制肿瘤生长
7. 类器官与PDX模型验证联合治疗效果
在人源胰腺癌类器官(PDOs)和患者来源异种移植(PDX)模型中,MEK抑制剂与脂肪酸氧化抑制剂联合治疗,均显著增强了ERK抑制剂的抗肿瘤效果,具有临床应用前景。
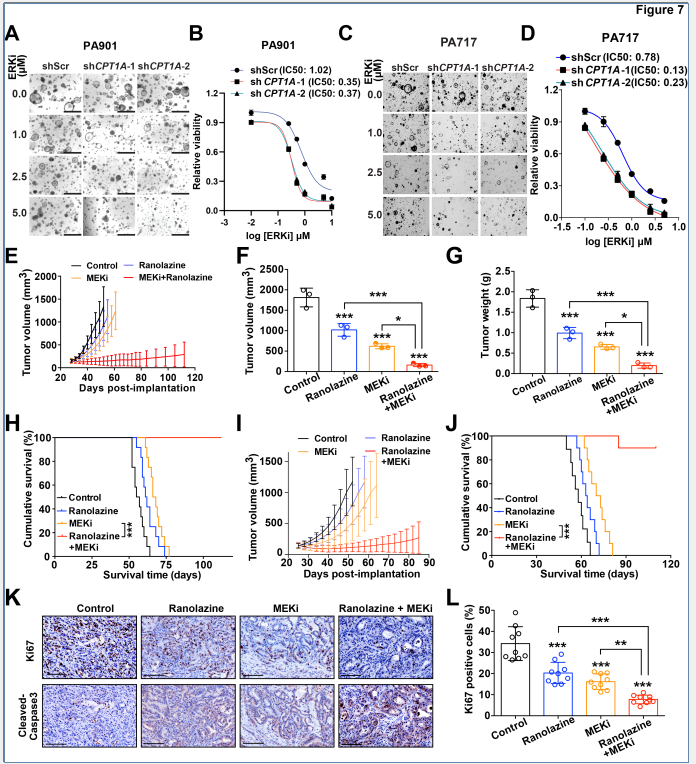
图7. 脂肪酸氧化抑制与MEK/ERK抑制剂联合使用在类器官和PDX模型中的效果
8. 免疫健全模型中KRAS/MEK/ERK联合脂肪酸氧化抑制
在免疫健全模型KPC1245中证实了MEK抑制剂与脂肪酸氧化抑制剂联合治疗的抗肿瘤治疗效果。脂质组学分析发现,KRASG12Di、MEKi和ERKi单独治疗均出现酰基肉碱积累和TG减少,而联合脂肪酸氧化抑制Ranolazine后TG水平恢复、酰基肉碱积累减少。
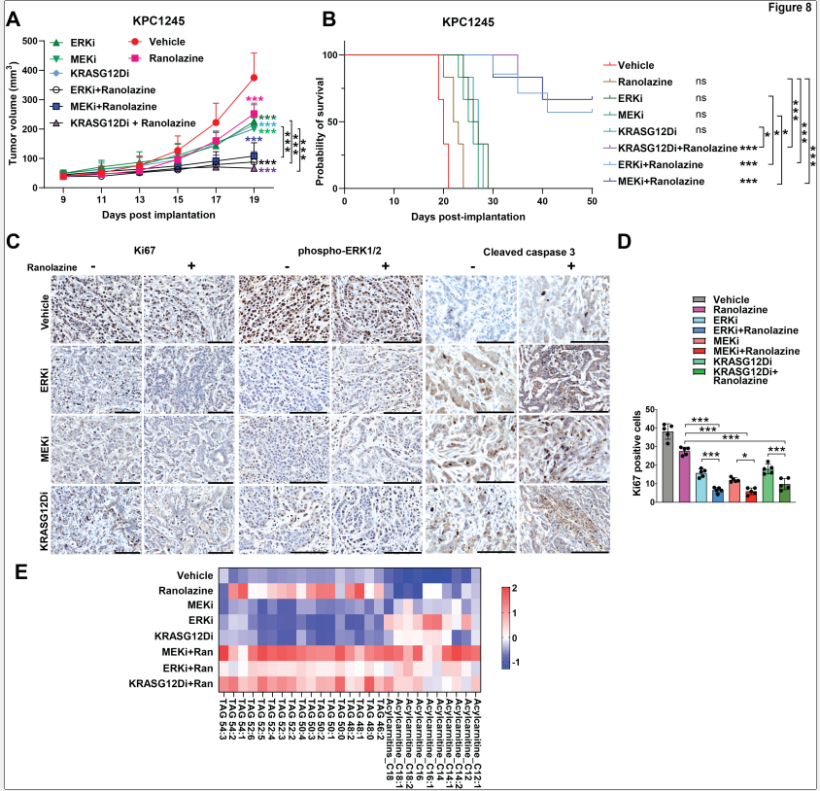
图8. 脂肪酸氧化抑制与KRAS/MEK/ERK抑制剂联合使用在免疫能力完整的模型中的效果
研究结论
这项研究不仅为靶向KRAS治疗胰腺癌耐药提供了重要理论依据,也为克服恶性肿瘤治疗耐药提供了具有直接临床转化潜力的联合用药方案。随着KRAS G12D抑制剂等新型靶向药物的临床推进,基于代谢脆弱性的联合治疗策略将为胰腺癌患者带来新的希望。
参考文献
Thakur R, et al. KRAS Signaling Inhibition Induces a Targetable Metabolic Dependency on Lipophagy-Dependent Fatty Acid Oxidation in Pancreatic Cancer. Cancer Res. 2026
扫描二维码阅读原文

绘谱帮你测
麦特绘谱开创性地搭建了医学领域高端代谢组学技术平台,覆盖了非靶向-全定量-代谢流等全方位的高端医学代谢组解决方案,同时全面布局微生物组学、转录组学和蛋白质组学等多组学技术服务,已成为全球多组学研究者的优选合作伙伴。麦特绘谱拥有QL1000、Q1000、Q500、Q300、Q200和胆汁酸、短链脂肪酸、色氨酸及吲哚衍生物、多胺和TMAO类等各类小分子代谢物、非靶向代谢组学和同位素示踪代谢流技术等共50+系列检测方法;已为数百家三甲医院、科研院所和企业提供多组学解决方案,协助客户与合作伙伴发表SCI文章600+篇,累计影响因子6000+,平均IF>10,包括Cell, Science, Nature, Cancer Cell, Signal Transduction and Targeted Therapy, Nature Biotechnology, Cell Metabolism等权威期刊。
