
文献解读
绘谱导读 | 顶刊拆解代谢免疫调控新机制:从肿瘤、代谢消化疾病到应激
本期导读汇总发表于Cell, Immunity, Gut, Nature系列子刊的顶尖研究,围绕肿瘤病变、代谢异常、机体应激三大方向:在肿瘤领域,棕榈酸重编程中性粒细胞、破坏血管完整性以促进乳腺癌肺转移;JOSD1通过调控PGAM1泛素化-乳酰化修饰转换加速肝癌恶性进展;DGAT1介导脂质代谢差异化,造成CD8⁺T细胞抗肿瘤反应的性别差异,而嘌呤补救通路可保护CD8⁺T细胞抵御代谢应激、维持抗肿瘤免疫。在代谢疾病研究中,二甲双胍依托抑制肠道上皮线粒体复合物Ⅰ实现血糖管控;B细胞缺失重塑肝脏谷氨酸代谢通路,直接限制机体运动耐力;肠道菌群代谢产物吲哚经由海马AhR通路,同步调控肠易激综合征的肠道与情绪病变。应激层面,外界热应激可通过皮肤-下丘脑神经轴扰乱全身代谢稳态,诱发代谢功能障碍。
导读目录
- Immunity | 棕榈酸重编程中性粒细胞,破坏血管完整性并促进乳腺癌肺转移
- Gut | JOSD1通过调节PGAM1上的泛素化-乳酰化转换来驱动肝细胞癌的恶性发展
- Cell | B细胞缺失通过重塑肝脏谷氨酸代谢限制运动耐力
- Cell Metabolism | 肠道微生物代谢物吲哚经海马AhR信号通路调控肠易激综合征的情绪症状
- Nature Metabolism | DGAT1介导CD8+ T细胞抗肿瘤反应的性别特异性
- Nature Metabolism | 二甲双胍通过抑制肠道上皮细胞的线粒体复合物I来促进血糖控制
- Cell | 热应激通过皮肤-下丘脑轴促进代谢功能障碍
- Nature Immunology | 嘌呤补救途径保护CD8+ T细胞免受代谢应激损伤
资源领取
本期导读文献原文,请在公众号后台回复“2026年5月绘谱导读”,即可获取资源链接。
一、Immunity | 棕榈酸重编程中性粒细胞,破坏血管完整性并促进乳腺癌肺转移

三阴性乳腺癌(TNBC)侵袭性强、复发率高、治疗手段有限,且易肺转移,显著影响患者预后。肺部在转移前微环境形成过程中,会为乳腺癌转移创造一个有利的微环境,其特征包括驻留细胞的代谢重编程、抑制性中性粒细胞的招募以及血管重塑。然而脂质在调控中性粒细胞与内皮细胞相互作用,尤其是促进肿瘤细胞外渗方面的作用仍不明确。本研究发现,TNBC诱导肺血管内皮细胞合成并释放大量 PA(棕榈酸),通过TLR4-NF-κB通路激活中性粒细胞分泌LCN2(脂质运载蛋白2),进而破坏肺血管屏障、促进肿瘤细胞转移定植,强调了代谢干预可能是针对乳腺癌肺转移的一种潜在治疗策略。
1. 通过对构建的4T1-GFP乳腺癌原位肺转移模型进行转录组与脂质组分析,发现脂质氧化和脂肪酸生物合成通路显著富集,游离脂肪酸和甘油三酯在肺组织中特异性升高。
2. 通过对长链脂肪酸定量检测,结果发现饱和脂肪酸和多不饱和脂肪酸均有所增加,其中棕榈酸的升高最为显著,体内外实验进一步证实,外源性补充棕榈酸不影响原位肿瘤生长,但可显著增强4T1、AT3细胞的肺转移能力。
3. 对肿瘤小鼠肺中性粒细胞进行转录组检测,发现多种炎症因子的表达水平显著升高,但只有LCN2明显破坏了内皮细胞的屏障完整性,进一步检测肺血管内皮细胞中紧密连接相关蛋白的表达,只有ZO-1显著减少。
4. 通过体外细胞实验表明,棕榈酸可直接激活中性粒细胞TLR4-MyD88-NF-κB信号轴,进而上调LCN2的表达与分泌。
5. 术后模型、化疗联合模型研究发现,临床降糖药GLP-1受体激动剂可抑制内皮脂肪酸合成,抑制肺转移,且与PD-1抑制剂具有协同治疗效果。
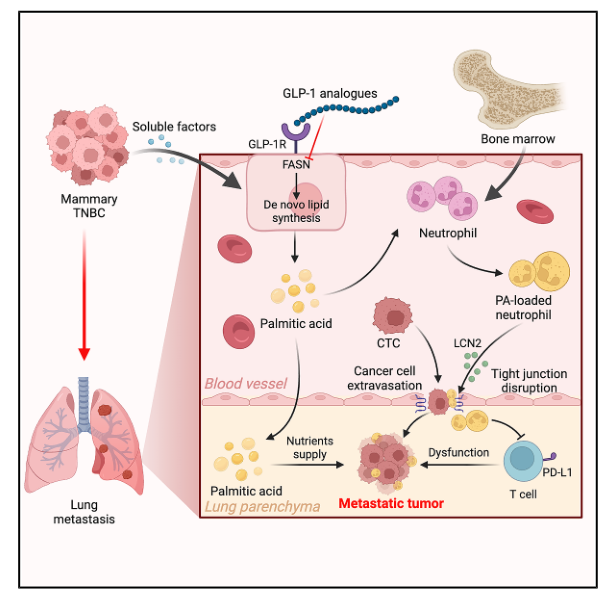
参考文献:Wei J W, Dong J, Zou Z Y, et al. Palmitic acid reprograms neutrophils to compromise vascular integrity and promote breast cancer lung metastasis. Immunity. 2026.
二、Gut | JOSD1通过调节PGAM1上的泛素化-乳酰化转换来驱动肝细胞癌的恶性发展

肝细胞癌(HCC)是全球第六常见的恶性肿瘤,也是导致癌症相关死亡的第三大原因,给全球健康带来了沉重的负担。代谢重编程是HCC的一个显著特征,它能促进肿瘤的快速生长并实现免疫逃逸。蛋白质翻译后修饰(PTM)的相互作用是细胞过程的关键调节因子,然而,其在HCC中对代谢重编程的贡献尚不清楚。本研究通过多组学分析以及细胞系、动物模型和患者样本中的功能及机制研究表明,JOSD1-AARS1轴调控了PGAM1上的泛素化-乳酰化相互作用,其中JOSD1起着关键的上游分子开关作用,能够驱动HCC中的代谢重编程和免疫逃逸。靶向JOSD1有望成为调节肿瘤代谢并提高免疫治疗效果的治疗策略。
1. 通过分析三个独立队列的转录组数据,发现碳水化合物代谢显示出最强的预后相关性,在与碳水化合物相关通路中,糖酵解表现出最强的富集和预后相关性,JOSD1是HCC糖酵解关键调控因子。体外和体内研究进一步表明,JOSD1能够促进肝癌细胞增殖、迁移和侵袭。
2. 通过蛋白质组分析发现,JOSD1显著上调糖酵解通路,不影响TCA循环。通过U-13C6-glucose代谢流实验,发现JOSD1过表达组中糖酵解代谢产物M3-乳酸的含量显著增加,而与三羧酸循环相关的代谢物则未显示出明显的变化。
3. 通过免疫沉淀-质谱技术以及泛素组学相关分析,发现糖酵解酶PGAM1是JOSD1的候选靶点,JOSD1通过去泛素化抑制PGAM1的蛋白酶体降解,关键位点为PGAM1-K251,且高度保守。
4. 通过PGAM1酶活检测以及Co-IP等实验进一步发现,AARS1是 PGAM1-K251的乳酸转移酶,JOSD1作为分子开关,启动AARS1介导的 PGAM1乳酸化。
5. 通过单药以及联合抗PD-1治疗发现,靶向抑制JOSD1与抗PD-1疗法可产生显著协同抗肿瘤效果,有效恢复CD8+细胞毒性T细胞的浸润与功能。
参考文献:Li Q, Yu K, Zhou S, et al. JOSD1 drives hepatocellular carcinoma malignancy by modulating the ubiquitination-lactylation switch on PGAM1. Gut. 2026.
三、Cell | B细胞缺失通过重塑肝脏谷氨酸代谢限制运动耐力

B细胞耗竭疗法已是B细胞淋巴瘤和部分自身免疫病的临床常规治疗手段,但B细胞是否具备超越体液免疫的生理性调控功能尚不清楚。运动能力依赖骨骼肌、心血管与代谢系统的复杂协同,而免疫-运动之间的调控机制仍是空白。本研究揭示了B细胞通过非免疫依赖方式调控运动能力的新功能,建立了“B细胞-TGF-β1-肝脏谷氨酸代谢-骨骼肌Ca²⁺信号与线粒体功能”的跨器官代谢调控轴,为理解运动能力的分子调控机制提供了新视角。
1. 通过行为学测试、代谢笼、透射电镜发现:B细胞缺失的小鼠运动耐力下降40%–50%,伴随骨骼肌线粒体数量减少、嵴结构受损,能量消耗降低且脂肪氧化偏好升高。
2. 代谢组学:采用非靶向代谢组学初筛差异代谢物,锁定氨基酸模块;再用靶向代谢组学精准定量谷氨酰胺(Gln)、谷氨酸(Glu)及相关代谢物,证实Glu含量是唯一在运动后小鼠肌肉中上升,而在血清中下降的氨基酸,其水平与B细胞数量呈正相关。
3. 同位素代谢流示踪实验:通过小鼠尾静脉恒速输注[U-¹³C₅]Gln发现,B细胞缺失的小鼠肝脏Gln_M5堆积和Glu_M5降低,提示Gln→Glu转化通量下降。
4. scRNA-seq、CUT&RUN及过继转移实验揭示分子轴:B细胞是外周TGF-β1主要来源,其通过SMAD2/3直接转录激活肝脏Gls2(催化Gln→Glu)和Slc7a5(介导Glu转运);肝细胞特异性双敲除这两个基因后,可完全重现 B细胞缺失所导致的运动能力下降表型。
5. 钙成像、Seahorse及干预实验验证下游机制:Glu通过增强肌管Ca²⁺振荡与线粒体呼吸恢复运动能力,Ca²⁺螯合剂可阻断该效应,确认钙信号为核心功能介质。
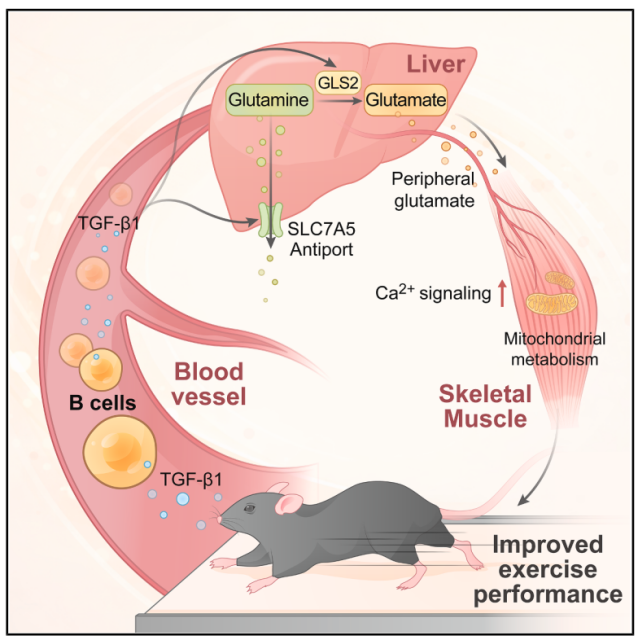
参考文献:Mao Y, et al. B cell deficiency limits exercise capacity by remodeling liver glutamate metabolism. Cell. 2026.
四、Cell Metabolism | 肠道微生物代谢物吲哚经海马AhR信号通路调控肠易激综合征的情绪症状

肠易激综合征(IBS)是一种与肠道微生物群改变相关的肠道疾病,常与情感障碍共病。肠道微生物群可通过多条相互关联的通路影响情绪,包括迷走神经介导的神经信号、炎症介导的免疫调节,以及微生物代谢物介导的代谢信号。本文旨以IBS为切入模型,鉴定驱动情绪紊乱的关键菌源代谢物及中枢机制,并评估其临床转化潜力。
1. 通过16S rRNA测序发现,IBS患者粪便中Alistipes属(另枝菌属)的整体丰度显著低于健康对照。进一步通过宏基因组在物种水平锁定菌种A. shahii(沙氏另枝杆菌)是IBS中下降最显著的物种之一,且其丰度与情绪量表得分高度相关。
2. 非靶向代谢组学发现IBS小鼠粪便代谢谱整体紊乱,筛选出吲哚是与A. shahii丰度以及情绪行为关联最强的候选代谢物。靶向色氨酸代谢组学进一步验证,吲哚在IBS小鼠及临床患者的粪便、血清中均显著降低,且与焦虑/抑郁评分呈显著负相关。
3. 宏基因组数据揭示IBS中tnaA基因丰度大幅下降,主要由A. shahii和Bacteroides的缺失导致。体外酶活实验证实,抑制TnAse会减少吲哚生成并诱发情绪症状,而回补TnAse则恢复表型。
4. 吲哚已知主要通过芳香烃受体AhR发挥作用,口服地奥司明能显著激活海马vDG区AhR信号,提升神经元活性,并在IBS小鼠及接受IBS菌群移植的无菌小鼠中均表现出确切的抗焦虑/抗抑郁效果。
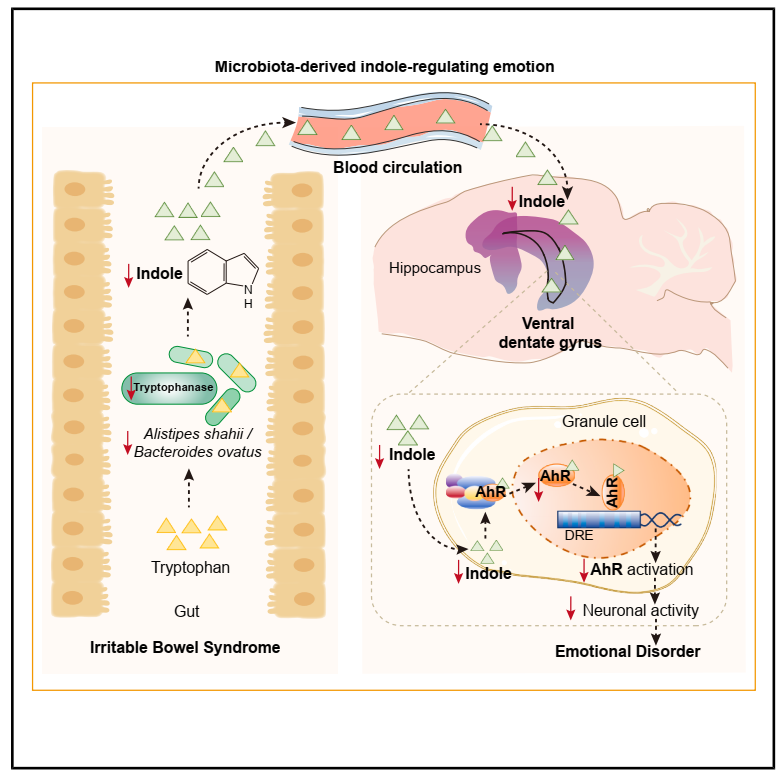
参考文献:Wang TT, et al. Gut microbial metabolism via hippocampal indole-AhR signaling regulates emotional symptoms. Cell Metab. 2026.
五、Nature Metabolism | DGAT1介导CD8+ T细胞抗肿瘤反应的性别特异性

DGAT1是催化甘油三酯(TAG)合成最后一步的关键酶,但其在CD8+ T细胞抗肿瘤免疫中的功能尚不明确。本研究发现,DGAT1在CD8+肿瘤浸润淋巴细胞中具有性别二态性作用。Dgat1缺失在雌性小鼠中增强抗肿瘤免疫;而在雄性小鼠中,Dgat1缺失则导致脂肪酸过氧化、内质网应激和Tex细胞死亡。上述效应由雄激素受体(AR)信号介导,敲除Ar或过表达GPX4可挽救Dgat1缺陷CD8+ T细胞的存活。
1. 首先通过分析小鼠B16模型及人类结直肠癌样本发现,DGAT1在CD8⁺ TILs中上调且与耗竭标志物共表达,是TAG合成的主要酶。进一步研究发现,T细胞特异性Dgat1敲除在雌性小鼠中抑制肿瘤、增加progenitor Tex细胞,在雄性小鼠中却促进肿瘤、减少progenitor Tex。过继转移实验证实,该性别二态性为CD8⁺ T细胞固有且依赖肿瘤微环境,而非系统性效应。
2. 靶向代谢组学分析显示,雄性小鼠肿瘤间质液(TIF)中 TAG 和甘油磷脂含量较高,但 PUFA-TAG 相对缺乏,LPE/PE比值较低。网络分析显示雄性TIF中TAG合成通路高度整合。相关性分析发现高甘油磷脂与雌性小肿瘤、雄性大肿瘤相关,提示雄性TIF存在独特的脂毒性压力。
3. 机制研究表明,雄性Dgat1 KO progenitor Tex细胞出现氧化应激和内质网应激相关细胞死亡特征,包括线粒体RNA富集、核糖体基因下调、内质网应激标志物上调及GPX4表达降低。体外实验进一步显示,睾酮可加重Dgat1 KO细胞的线粒体功能障碍、ROS与脂质过氧化物累积及内质网结构异常。
4. 为验证AR信号的关键作用,构建Dgat1与Ar双敲除P14细胞,过继转移至雄性宿主可完全逆转Dgat1单敲除所致的肿瘤快速生长并恢复progenitor Tex比例,但同时抑制向效应细胞分化。
5. 最后通过功能挽救实验证实,在Dgat1 KO细胞中过表达GPX4(抗铁死亡)或敲除Ddit3(介导内质网应激凋亡),均可恢复雄性宿主中Dgat1缺陷CD8⁺ T细胞的抗肿瘤功能并增加progenitor Tex比例,证明DGAT1通过清除脂质过氧化物、抑制内质网应激,保护AR信号下的CD8⁺ T细胞。
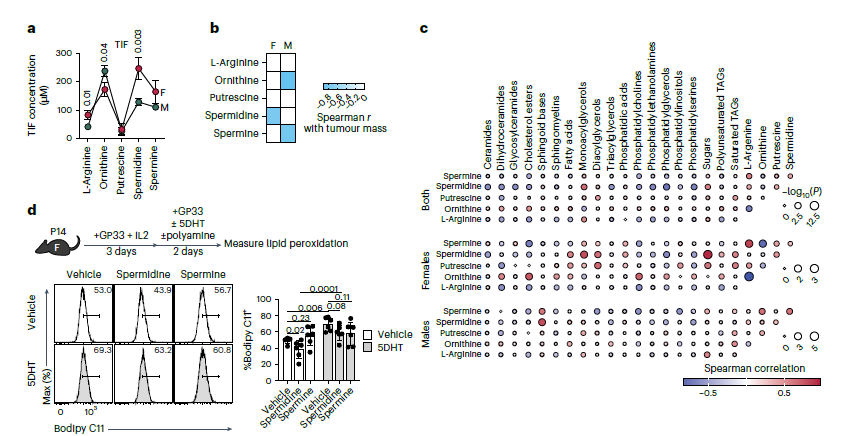
参考文献:Madi A, Shi H, Su M, et al. DGAT1 mediates sex-specific CD8+ T cell antitumour responses. Nat Metab. 2026.
六、Nature Metabolism | 二甲双胍通过抑制肠道上皮细胞的线粒体复合物I来促进血糖控制

二甲双胍是治疗2型糖尿病的一线药物,但其作用机制长期存在争议。传统观点认为其通过抑制肝脏糖异生发挥降糖作用,然而临床药理浓度下药物主要在肠道蓄积。本研究通过特异性敲除肠道上皮细胞线粒体复合物I,发现二甲双胍可通过抑制肠道复合物 I 促进葡萄糖清除,同时阐明了苯乙双胍和小檗碱共享这一作用通路。
1. 首先利用公开人类代谢组数据和基因工程小鼠模型,发现二甲双胍显著降低血浆瓜氨酸水平,该代谢物仅由小肠线粒体合成。通过构建肠道特异性表达酵母NDI1(抗二甲双胍的复合物I替代酶)的转基因小鼠(Vil-Cre:NDI1),发现二甲双胍降低空肠琥珀酸/富马酸比值的作用被完全阻断,直接证明其在体抑制肠道复合物I。
2. 随后通过FDG-PET和2-脱氧葡萄糖摄取实验评估肠道葡萄糖处置能力。结果显示二甲双胍显著增加对照小鼠肠道FDG和2DG6P积累,而在Vil-Cre:NDI1小鼠中该效应消失。同时,二甲双胍升高血乳酸、乳酰苯丙氨酸及GDF15的作用均依赖肠道复合物I抑制,提示该机制驱动肠道转变为葡萄糖代谢池。
3. 口服和腹腔葡萄糖耐量试验显示,在正常饮食和高脂诱导肥胖小鼠中,NDI1表达均显著削弱了二甲双胍改善糖耐量的能力,且对餐后血糖和胰岛素的抑制效应也部分丧失。此外,慢性饮水给药无效,而每日灌胃的重复脉冲暴露才是疗效关键,非累积性稳态改变。
4. 通过丙酮酸耐量试验评估对糖异生的影响。二甲双胍改善丙酮酸耐量并升高血乳酸的作用在Vil-Cre:NDI1小鼠中显著减弱,而肝脏代谢组无变化,说明药物通过肠道复合物I抑制重定向丙酮酸流向肠道进行糖酵解,而非直接抑制肝脏糖异生。
5. 最后,苯乙双胍降低血糖的作用在Vil-Cre:NDI1小鼠中显著减弱。小檗碱联合肠道P-糖蛋白抑制剂恩曲他滨后,其降糖作用完全依赖于肠道复合物I抑制,证实该机制是多种天然及合成降糖化合物的共同治疗靶点。
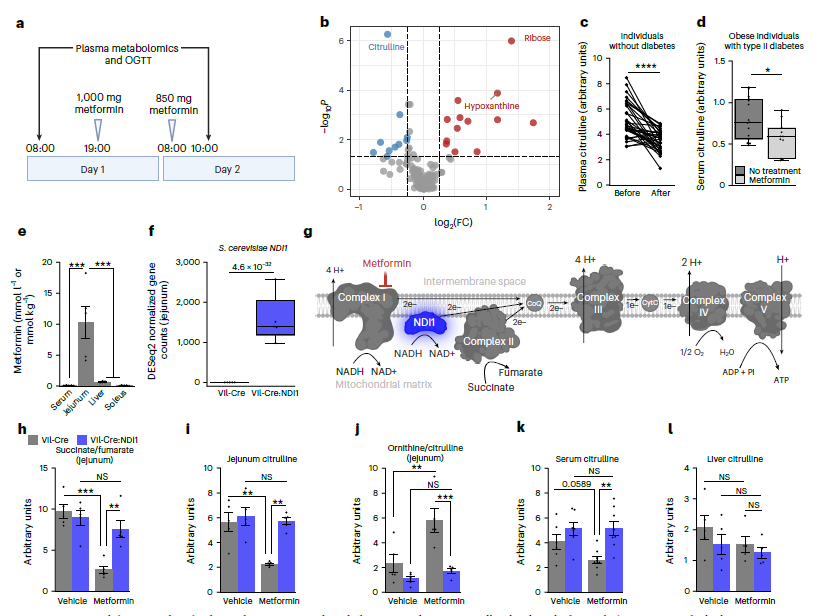
参考文献:Sebo ZL, Chakrabarty RP, Grant RA, et al. Metformin inhibits mitochondrial complex I in intestinal epithelium to promote glycemic control. bioRxiv [Preprint]. Nat Metab. 2026.
七、Cell | 热应激通过皮肤-下丘脑轴促进代谢功能障碍

热应激是否对代谢健康产生持久影响尚不明确。本研究发现热应激小鼠在随后高脂饮食下更易出现代谢功能障碍。热应激诱导皮肤来源的KLK14升高,通过受体CHL1印记下丘脑PVN LRRC7⁺星形胶质细胞,经ALKBH1介导的N6-mA去甲基化表观遗传修饰上调MAOB表达,促进GABA合成,抑制PVN OXT神经元活性,经交感神经系统降低脂肪脂解,驱动内脏脂肪沉积。维生素A可减少KLK14产生并改善热应激相关代谢障碍。
1. 首先,构建热应激小鼠模型 (37°C,6h/天,1周),经2周静息期后饲喂高脂饮食,发现热应激小鼠出现体重增加、糖耐量受损、胰岛素抵抗及内脏脂肪蓄积,伴脂肪脂解和脂肪酸氧化通路抑制,该效应4周后仍持续,表明热应激赋予持久代谢印记。人体外卖骑手队列显示累积高温暴露与体重、BMI和腹内脂肪面积长期增加正相关。
2. 进一步研究发现,下丘脑LRRC7⁺星形胶质细胞在热应激后显著扩增并富集于PVN。ATAC-seq揭示热应激诱导的染色质开放区在静息期后仍持续,形成”热记忆”。过继转移实验证实LRRC7⁺星形胶质细胞可独立驱动代谢障碍,化学遗传学抑制则可逆转。
3. 利用血清蛋白质组学鉴定出皮肤来源的KLK14为关键循环因子。热应激选择性升高皮肤角质细胞KLK14表达,静脉注射rKLK14可复现LRRC7⁺星形胶质细胞扩增,皮肤特异性Klk14敲除则阻断热记忆。KLK14经受体CHL1作用于LRRC7⁺星形胶质细胞,通过ALKBH1介导N6-mA去甲基化上调MAOB,促进GABA合成,抑制PVN OXT神经元,经交感神经系统减少脂肪去甲肾上腺素释放与脂解。
4. 最后,发现维生素A可抑制皮肤KLK14产生,同时降低血清KLK14水平,减少LRRC7⁺星形胶质细胞扩增,改善小鼠热记忆诱导的代谢障碍。人体随机对照试验中,维生素A补充(5000 IU/天)显著降低骑手血清KLK14水平,减少BMI、腰围、腹内脂肪面积和胰岛素抵抗指数增幅,证实维生素A对热应激代谢损伤的保护作用。
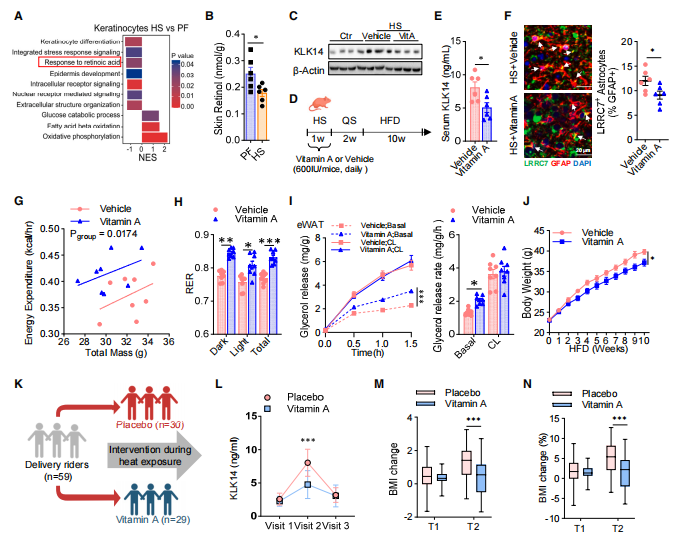
参考文献:Zhou HY, Feng X, Wen J, et al. A skin-hypothalamus axis couples heat stress and metabolic dysfunction. Cell. 2026
八、Nature Immunology | 嘌呤补救途径保护CD8+ T细胞免受代谢应激损伤

高脂饮食 (HFD) 引起的代谢应激可通过持久的代谢重编程损害抗肿瘤免疫。本研究发现,即使仅短暂暴露于HFD (6周) 后恢复正常饮食10周,CD8+ T细胞的抗肿瘤功能仍显著受损。机制上,HFD导致CD8+ T细胞脂质组和代谢组发生持久改变——多不饱和脂肪酸 (PUFA) 富集的磷脂酰胆碱和磷脂酰乙醇胺显著积累,同时抗氧化代谢物 (如谷胱甘肽) 耗竭,使细胞对铁死亡 (ferroptosis) 产生内在脆弱性。在氧化应激下,CD8+ T细胞可利用黄嘌呤 (xanthine)补救途径生成GTP,进而增强四氢生物蝶呤 (BH4)合成。补充xanthine可降低肿瘤引流淋巴结中的脂质过氧化,恢复曾接受HFD小鼠的抗肿瘤免疫。
1. 首先,将小鼠分为正常饮食 (ND)、短期HFD后切换 (SD) 和持续HFD三组,结合LLC肺癌模型评估抗肿瘤免疫,发现SD组肿瘤生长速度与持续HFD组相似,均显著快于ND组。多组学分析揭示,HFD在CD8+ T细胞中诱导了持久代谢重编程:PUFA富集磷脂显著积累,抗氧化剂谷胱甘肽等持续耗竭,脂质代谢基因Acsl4、Lpcat3表达上调。这些变化在恢复正常饮食10周后仍不逆转,提示 CD8⁺ T 细胞存在饮食诱导的”代谢记忆”效应。
2. 进一步发现,CD8+ T细胞在氧化应激下特异性激活黄嘌呤补救途径:xanthine经HPRT1催化依次转化为次黄嘌呤、IMP、GMP,最终生成GTP;GTP进一步合成BH4。BH4作为强效抗氧化剂,直接清除脂质过氧自由基,抑制铁死亡。CD8+ T细胞对xanthine的摄取显著高于CD4+ T细胞和B细胞,人类PBMC实验也验证了该保护作用的保守性。
3. 机制研究表明,HFD来源的 CD8+ T细胞表现出对铁死亡的内在脆弱性。RSL3 (GPX4抑制剂) 处理下,HFD/SD组CD8+ T细胞死亡显著增加;ACSL4表达上调,PUFA富集磷脂作为铁死亡底物大量积累。肿瘤引流淋巴结中脂质过氧化产物增加而抗氧化代谢物降低,体内铁死亡压力加剧。补充xanthine或biopterin可有效抑制铁死亡,恢复CD8+ T细胞存活及抗肿瘤功能。Rag2-/-小鼠实验排除xanthine对肿瘤细胞的直接作用,确证其通过保护CD8+ T细胞发挥作用。
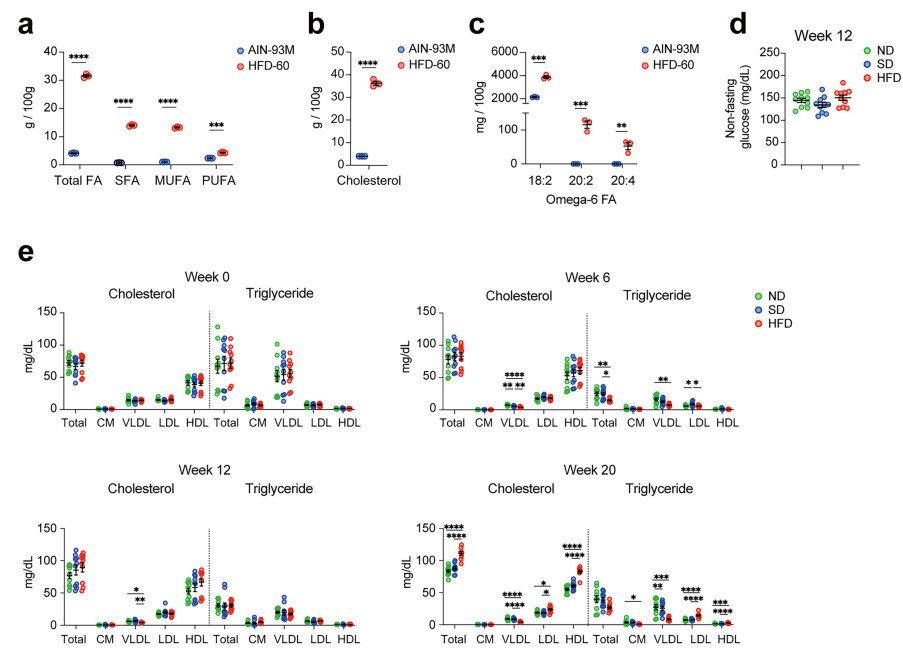
参考文献:Tajima, M., Hao, H., Zhang, B. et al. Purine salvage pathway protects CD8+ T cells from metabolic stress. Nat Immunol. 2026.
