
文献解读
Immunity | lnc-Dpf3抑制CCR7介导的树突状细胞迁移
海军军医大学曹雪涛研究团队通过系列研究证明长链非编码RNA(lnc-Dpf3)直接抑制HIF -1α依赖的糖酵解来抑制CCR7介导的树突状细胞(DC)迁移,以避免异常的炎症反应。
表观遗传途径在生理过程调节中具有重要作用,长链非编码RNA(lncRNA)以谱系或信号依赖等方式调节免疫应答,而探索lncRNA如何与代谢途径相互作用来调节炎症反应,对深入理解免疫和炎症的分子机制至关重要。外周未成熟DCs在微生物产物或炎症信号的驱动下成熟,CC-趋化因子受体7 (CCR7)水平上调,CCR7的配体CCL21和CCL19将控制DC向引流淋巴结(dLNs)转运以诱导适应性免疫,异常的DC转运和积聚与各种炎症性疾病的发病机制有关。加速DC迁移是快速诱导免疫系统消灭入侵病原体的必要条件,而及时终止DC迁移则是预防不必要炎症的必要条件。以往研究主要集中在增强CCR7在免疫反应早期触发DC迁移的阳性介质上,因此迫切需要在炎症后期及时识别抑制CCR7介导的DC迁移的调控网络。
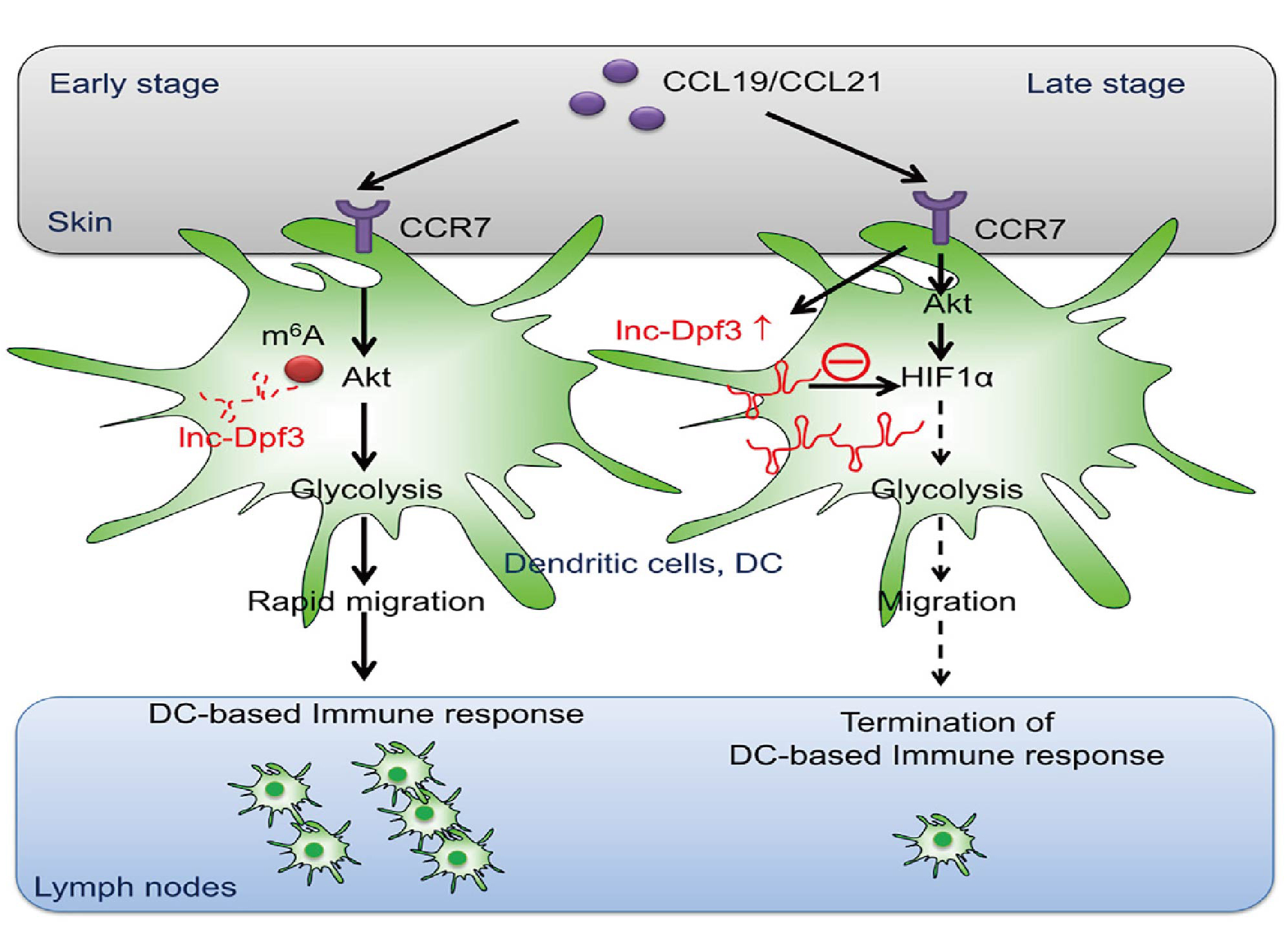
lnc-Dpf3抑制CCR7介导的DC迁移
基于已有研究基础,作者假设CCR7刺激可能会在DC向dLNs迁移高峰后(体内感染或刺激后约18-24小时)诱导细胞内的改变,以防止DC过多积累。采用LPS刺激的骨髓源性DC(成熟DCs, mDCs),在体外用CCR7配体(CCL19, CCL21)刺激mDCs 12小时,通过lncRNA谱检测共筛选到34个lncRNA在CCR7刺激的mDCs中存在差异表达,qRT-PCR分析证实了18个上调lncRNA 中的6个在CCR7刺激后显著上调(图1B)。通过对这6种lncRNA进行RNA干扰(RNAi)介导的功能筛选,发现CCR7介导的mDC体外迁移可以通过沉默内含子lncRNA(n274819,位于小鼠Dpf3基因内含子1内<被命名为lnc-Dpf3>)而显著增加(图1C)。lnc-Dpf3在脾脏CD11c+DCs表达量高于B细胞、T细胞、NK细胞。不同沉默剂沉默lnc-Dpf3均显著增加了CCR7介导的mDC迁移(图1D)。这些数据表明lnc-Dpf3在体外参与DC迁移抑制过程。
DC特异性lnc-Dpf3缺陷可选择性促进CCR7介导的DC迁移
作者通过同源重组获得CD11c+ DC中lnc-Dpf3特异性缺失的lnc-Dpf3fl/flItgax-cre+小鼠以确定lnc-Dpf3缺失是否影响CCR7触发的DC迁移。研究结果发现lnc-Dpf3fl/flItgax-cre+小鼠的未成熟DC和mDCs在CCR7刺激12小时后都比同窝其它幼鼠的DC迁移得更多(图1E)。腺病毒介导的lnc-Dpf3过表达使得12小时后CCR7触发的DC迁移减少(图1F)。但lnc-Dpf3fl/flItgax-cre+小鼠的DC在CCR7刺激后的前4h内DC迁移速度没有发生变化,表明lnc-Dpf3在CCR7刺激后的早期不影响DC迁移。此外,体外刺激CCR7 12 h后lnc-Dpf3表达持续上调,提示lnc-Dpf3在体外选择性抑制CCR7诱导的晚期DC迁移。
在体内模型中,进一步探讨lnc-Dpf3在CCR7介导的DC迁移中的作用。与对照组mDCs相比, lnc-Dpf3沉默后的mDCs在注射后期(24-48h)向dLNs的迁移增加(图1G, 1H)。同样地,lnc-Dpf3fl/flItgax-cre+小鼠的mDCs在注射24-48小时后向dLNs的迁移也增加(图1I和S3E)。FITC(荧光素异硫氰酸酯)涂皮48 h后 lnc-Dpf3fl/flItgax-cre+小鼠dLNs中FITC+ DCs的比例增加(图1J和S3F)。因此,lnc-Dpf3可在体内抑制晚期DC向dLNs的迁移。在lnc-Dpf3fl/flItgax-cre+小鼠中,只有CCR7依赖的迁移DC亚群显示FITC表达增加(图1K和S3G),而独立于CCR7的迁移DC亚群并没有。在dLNs中,FITC+ DC的lnc-Dpf3表达水平明显高于FITC- DC(图S3I)。综上,体内和体外研究表明,lnc-Dpf3在CCR7诱导的DC迁移过程中表达上调,随后抑制DC迁移。
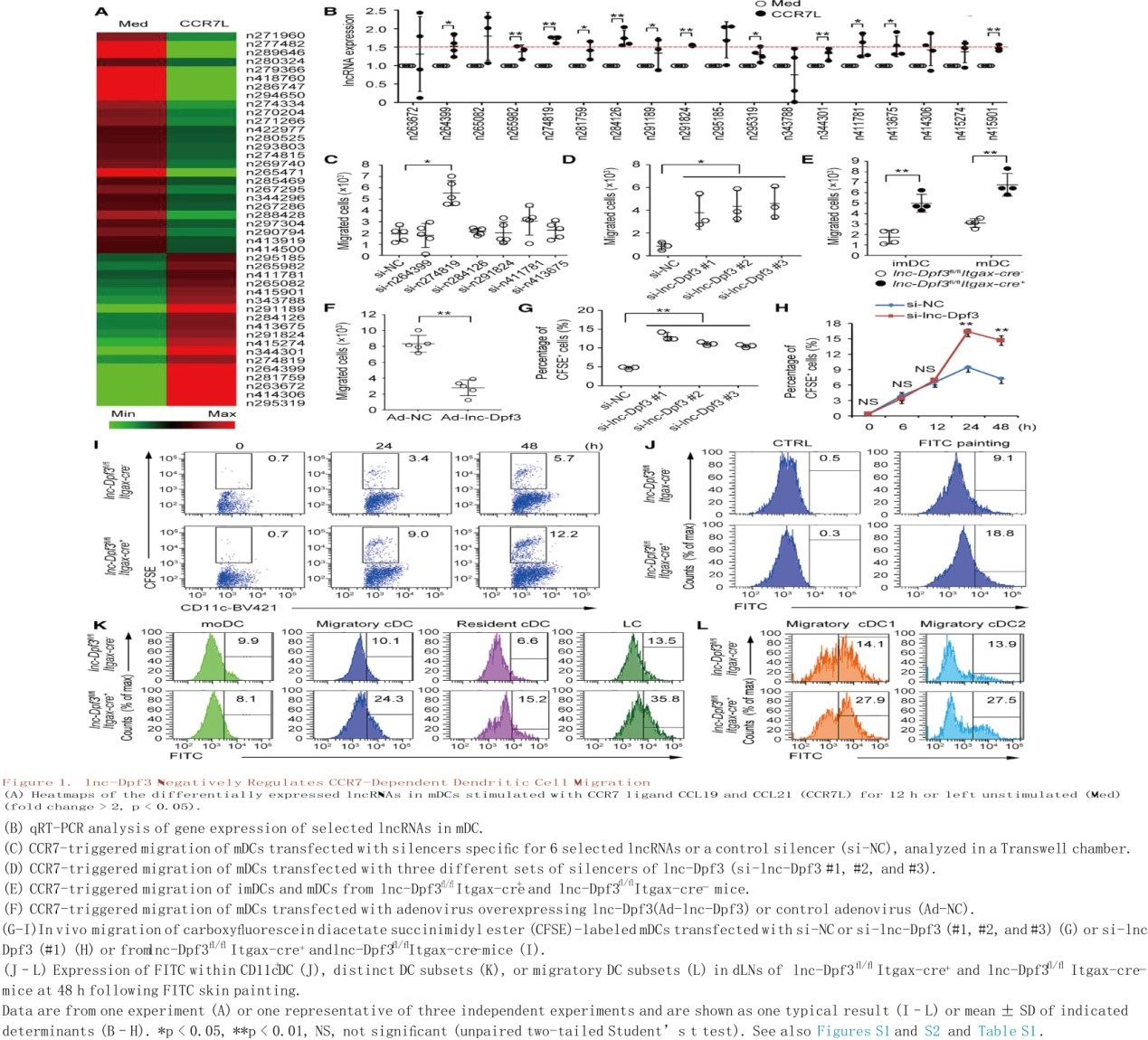
DC特异性lnc- dpf3缺陷小鼠的炎症反应更为严重
选取2,4-二硝基氟苯(DNFB)作为抗原的小鼠接触性过敏(CHS)皮炎模型以探讨体内lnc-Dpf3控制DC依赖性炎症反应中的生理作用。发现lnc-Dpf3fl/flItgax-cre+小鼠在CHS诱导后出现更严重的炎症反应,如耳肿胀程度加重、组织损伤和炎症浸润加重(图2A, 2B, S3J, S3K)。与同窝小鼠相比,体外DNBS再刺激后,lnc-Dpf3fl/flItgax-cre+小鼠T helper 1 (Th1)和Th17细胞数量增加,dLNs中干扰素(IFN)-γ和白细胞介素(IL)-17水平升高(图2C, 2D)。此外,接受DNBS负载的lnc-Dpf3沉默mDC小鼠在DNFB暴露后比注射负载DNBS的对照mDC小鼠产生更强的CHS应答(图2E-2G)。因此,DC中的lnc-Dpf3减弱了适应性反应和炎性损伤。此外,与同窝小鼠相比,lnc-Dpf3fl/flItgax-cre+小鼠在DNFB刺激后cDC1和cDC2向dLNs迁移增强,而在稳态条件下并没有(图2H)。在体内平衡和炎症条件下,dLNs中的迁移cDC1比非迁移cDC1的lnc-Dpf3表达更高(图2I)。因此,尽管在稳态和炎症条件下迁移DCs的lnc-Dpf3表达均上调,但在炎症条件下选择性减弱了CCR7介导的DC迁移。
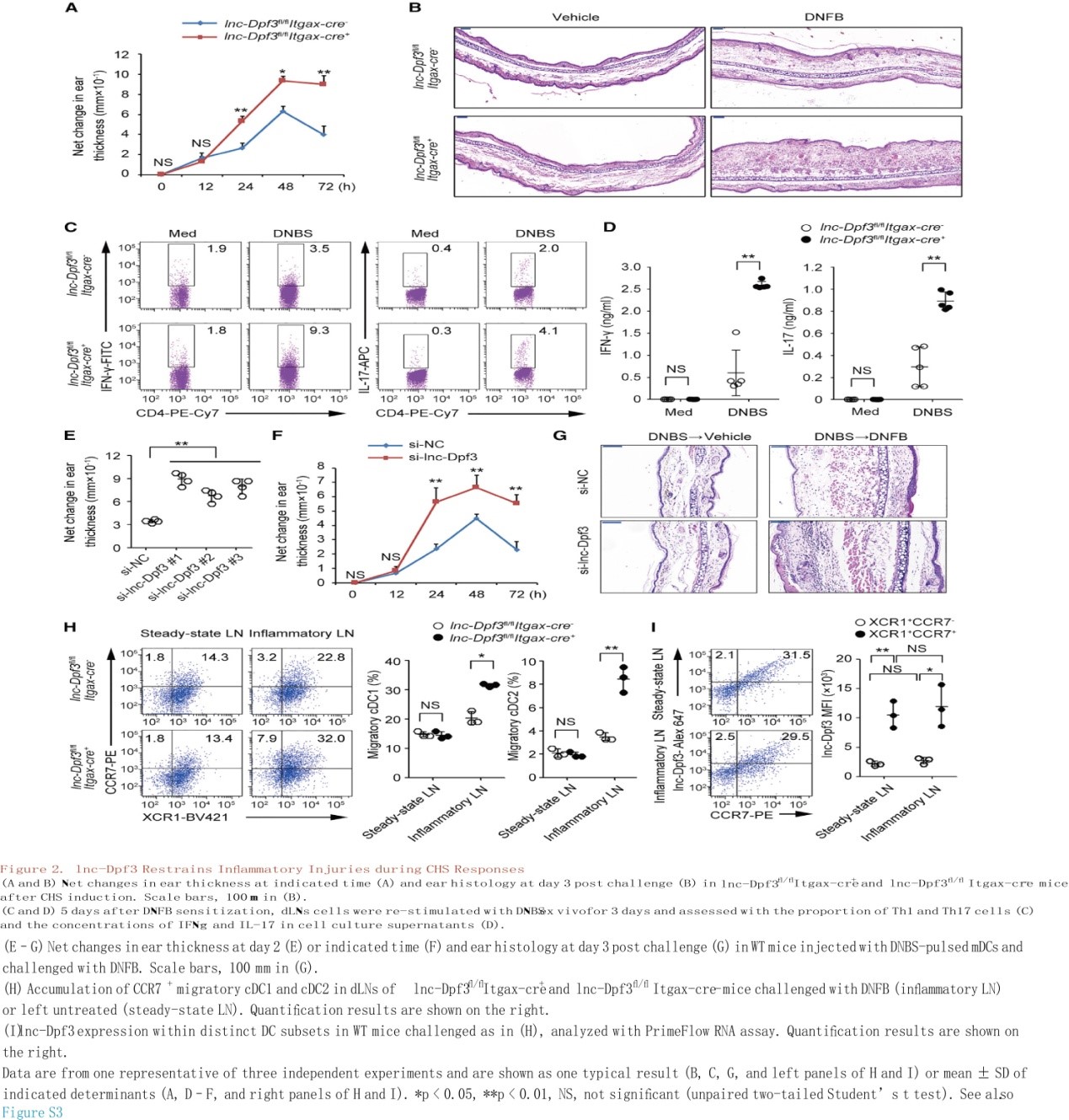
CCR7刺激通过去除m6A修饰上调lnc-Dpf3的表达,以防止其降解
作者首先证实CCR7并没有通过转录机制增加lnc-Dpf3的表达,进而探究CCR7刺激是否影响lnc-Dpf3的亚细胞定位。通过单分子RNA荧光原位杂交(FISH)提示lnc-Dpf3主要在胞质中,少部分位于细胞核中(图3A, 3B)。可见CCR7刺激对lnc-Dpf3的亚细胞分布没有明显影响(图3A)。因此,猜想CCR7刺激是否通过转录后调控RNA稳定性来增加lnc-Dpf3的表达。通过m6A RNA免疫沉淀(Me-RIP)在lnc-Dpf3上检测m6A修饰,在休眠的mDCs中检测到lnc-Dpf3的第22个片段显著富集了m6A,而CCR7刺激显著降低了m6A的峰值(图3C)。RIP测定显示lnc-Dpf3被Ythdf2(YTH N6-甲基腺苷RNA结合蛋白2)识别,而不是Ythdf1,并且Ythdf2对lnc-Dpf3的识别也被CCR7刺激下调(图3D)。Ythdf1或Ythdf2本身的表达不受CCR7刺激的影响(图3E)。此外,用siRNA沉默Ythdf2可上调lnc-Dpf3表达(图3F)。可见Ythdf2结合可加速甲基化转录物的降解。lnc-Dpf3共有14个m6A修饰位点(GGAC),其中2个位于第22片段,即3812- 3815 nt和3852 -3855 nt。通过质粒过表达野生型(WT)lnc-Dpf3和突变型(GGGC)(Dm6A-1, Dm6A-2),发现突变型导致lnc-Dpf3寿命延长(图3G)。Me-RIP证实了lnc-Dpf3高丰度的m6A修饰在突变型中降低,且在Dm6A-1+2突变中降低最明显(图3H)。同样地,lnc-Dpf3表达在突变型中增加,且在Dm6A-1+2突变中增加最明显,暗示了两个位点在介导lnc-Dpf3 RNA降解中发挥协同作用。总的来说,lnc-Dpf3在3812- 3815 nt和3852 -3855 nt处有m6A修饰,它们被Ythdf2识别并针对性降解。CCR7刺激通过介导lnc-Dpf3的m6A去甲基化,减轻依赖于m6A的RNA降解,从而上调lnc-Dpf3的表达。
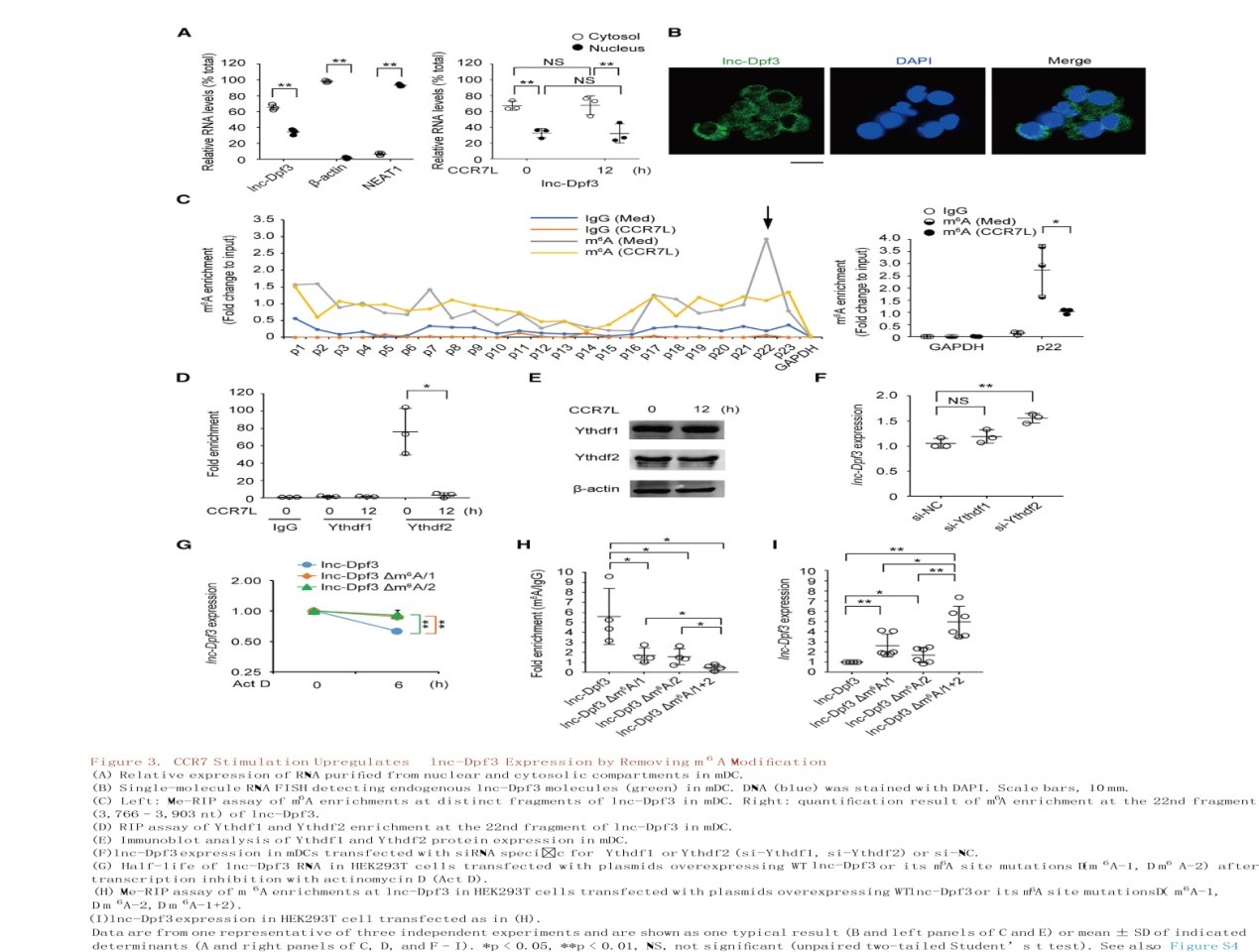
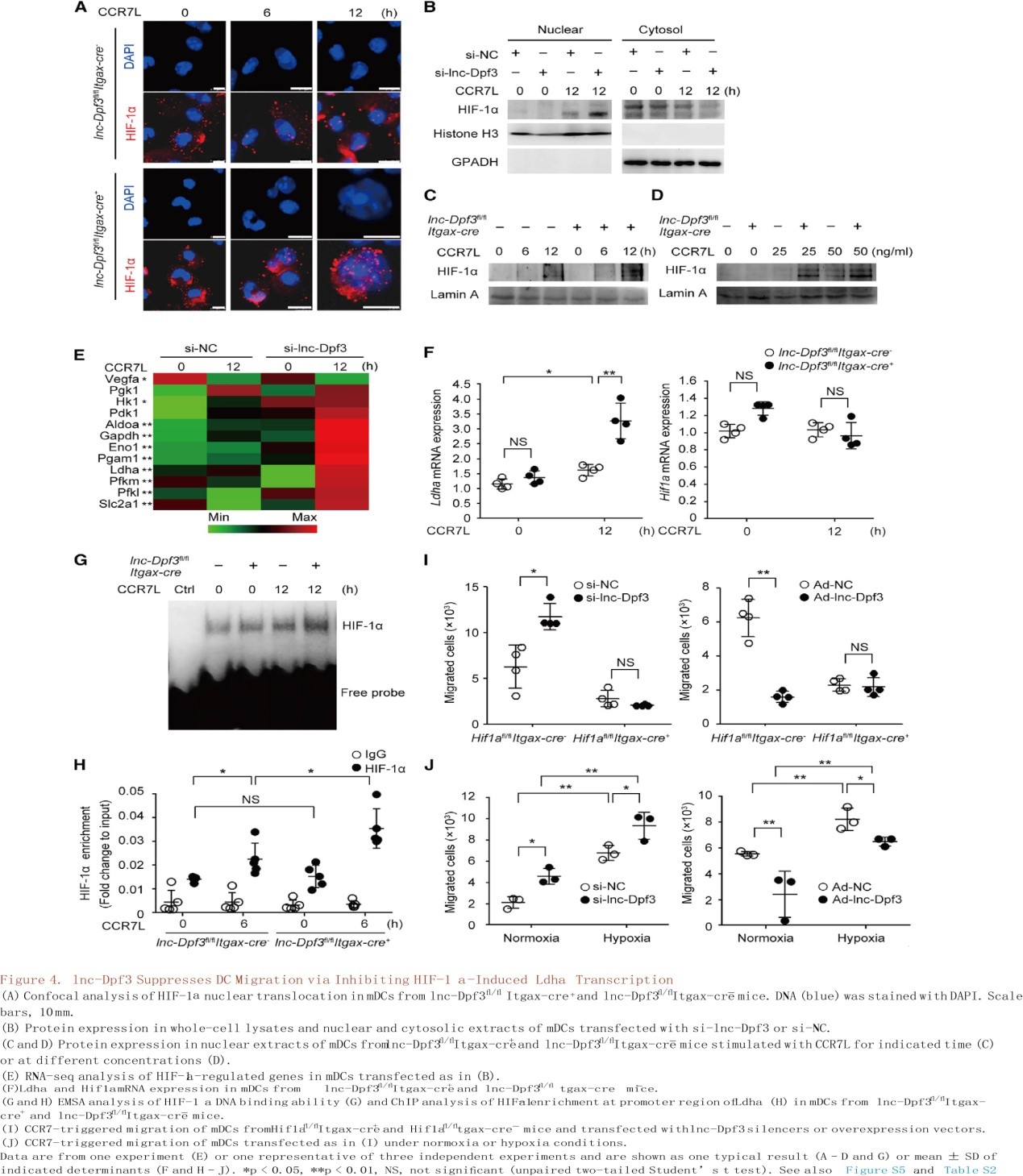
CCR7诱导的lnc-Dpf3通过靶向HIF-1α抑制DC迁移
在参与DCs先天激活的信号通路中,抑制Akt或其激活因子PI3K或其下游转录因子HIF-1α都可以显著阻断CCR7依赖的DC迁移。HIF-1α是细胞对低氧应激反应的中心转录因子,在先天性和适应性免疫反应中发挥重要作用。考虑到被感染组织的缺氧微环境,作者推测lnc-Dpf3可能通过调节HIF-1α的激活来调控CCR7介导的DC迁移。体内外研究表明DC特异性HIF-1α缺陷小鼠(Hif1afl/flItgax-cre+)DC迁移均受损(S4J-4M),可见CCR7介导的DC迁移需要HIF-1α。mDCs中lnc-Dpf3沉默或缺失增加了CCR7刺激诱导的HIF-1α入核,而不影响细胞内HIF-1α总浓度(图4A-4D)。RNA-seq分析证实,相比于正常DCs, lnc-Dpf3沉默的DCs中HIF-1α靶向的糖酵解基因上调,如HK1(己糖激酶)、Ldha(乳酸脱氢酶A)和Slc2a1(图4E)。qRT-PCR证实了lnc-Dpf3沉默或缺失的DCs中CCR7诱导的糖酵解基因Ldha显著增加(图4F, S5B, S5C)。通过电泳迁移率转移实验(EMSA)和芯片分析(图4G-H和S5F-G)确定mDCs中lnc-Dpf3沉默或缺失增加了Ldha基因启动子区域的HIF-1α DNA结合活性和积累。这些数据表明,lnc-Dpf3能够减弱CCR7诱导的HIF-1α活性和糖酵解基因Ldha的表达。更重要的是,HIF-1α缺乏可以阻断lnc-Dpf3沉默或过表达在增加或减少DC迁移中的作用(图4I)。相反,缺氧可促进CCR7介导的DC迁移并恢复lnc-Dpf3过表达DCs中受损的DC迁移(图4J)。总之,lnc-Dpf3通过靶向HIF-1α抑制CCR7介导的DC迁移。
lnc-Dpf3通过抑制HIF-1α依赖性糖酵解抑制DC迁移
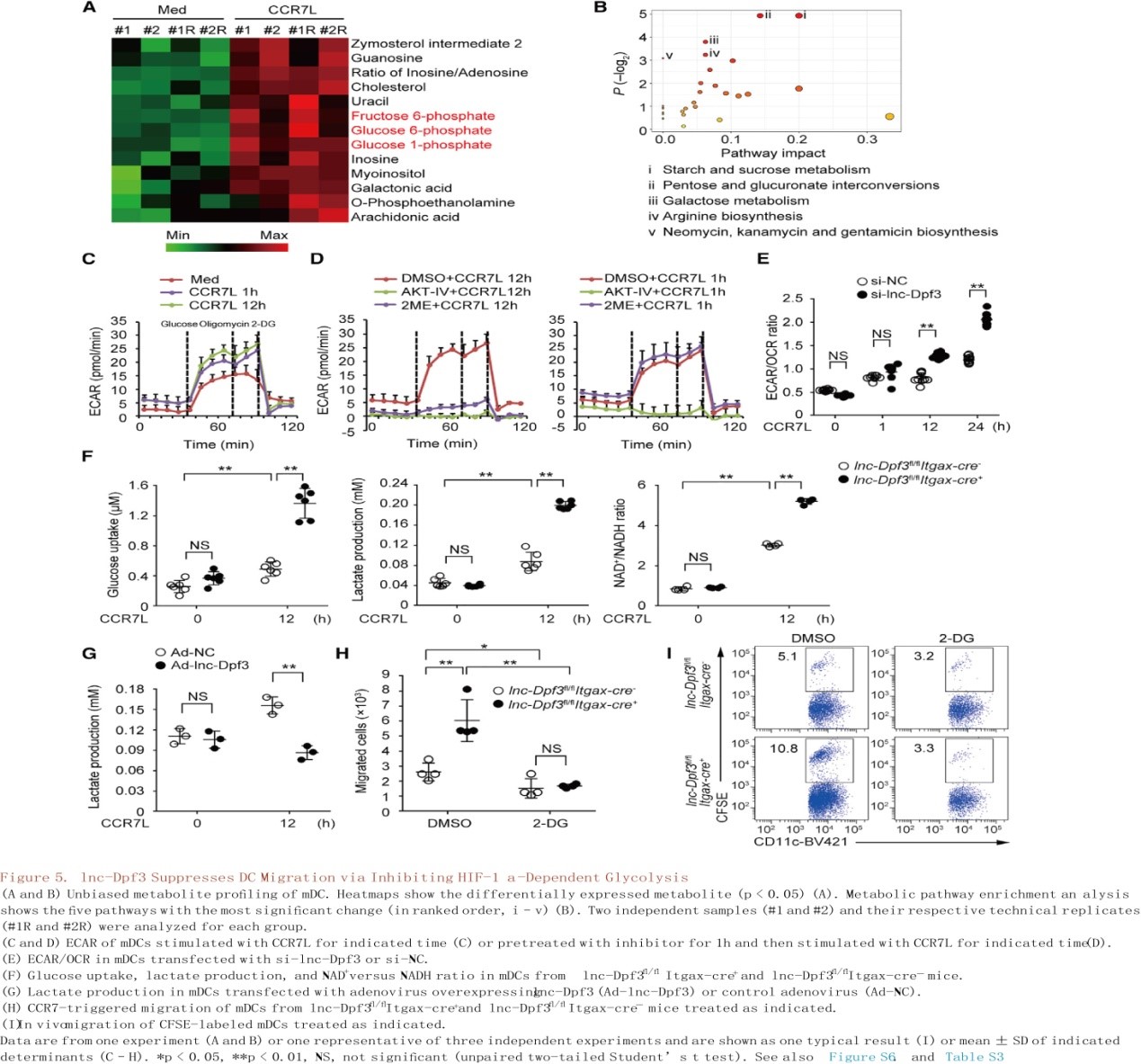
与RT-PCR和RNA-seq检测到的糖酵解基因的增加相一致,在CCR7刺激12小时后,mDCs非靶向代谢组学结果显示选择性CCR7诱导的葡萄糖代谢相关的中间体出现积累,如葡萄糖1-磷酸、葡萄糖6-磷酸和果糖6-磷酸(图5A;表S3)。同时分析了细胞外酸化(ECAR)和氧消耗(OCR)的速率,分别作为CCR7刺激后mDC中乳酸产生和线粒体能力的度量,ECAR / OCR比率可作为代谢向有氧糖酵解转变的度量。作者观察到CCR7刺激后DC的ECAR快速且持续增加(图5C)。尽管抑制Akt能够在早期和晚期都阻断糖酵解的增加,而HIF-1α能够选择性地阻断刺激后期糖酵解的爆发,提示HIF-1α对于CCR7诱导的晚期糖酵解尤为重要(图5D)。CCR7刺激后,lnc-Dpf3沉默DC中ECAR/OCR比值持续增加,且在12h后显著高于对照组(图5E)。lnc-Dpf3沉默或缺失的mDCs中葡萄糖摄取、乳酸产量和烟酰胺腺嘌呤二核苷酸(NAD+)与还原型(NADH)比值增加(图5F)。
相反,过表达lnc-Dpf3或DC特异性HIF-1α缺乏可抑制CCR7刺激的mDC中乳酸产生(图5G和S6D)。用糖酵解抑制剂2-脱氧葡萄糖(2-DG)处理DC可以抑制CCR7介导的DC迁移并且在体内外(图5H-I和S6F-G)消除lnc-Dpf3沉默或缺乏在增加DC迁移中的作用。糖酵解抑制可以抑制体内DC向dLNs迁移的早期和晚期,这说明糖酵解是CCR7依赖的DC迁移整个过程所必需的(图S6H)。因此,lnc-Dpf3通过抑制糖酵解来负向调控刺激后期DC迁移。综上所述,CCR7刺激可诱导DC中HIF-1α依赖的糖酵解反应,并伴随线粒体呼吸改变,而lnc-Dpf3通过抑制HIF-1α依赖的代谢重编程向有氧糖酵解转变,抑制CCR7介导的DC迁移。
lnc-Dpf3通过HRE基序直接抑制HIF-1α以抑制DC糖酵解
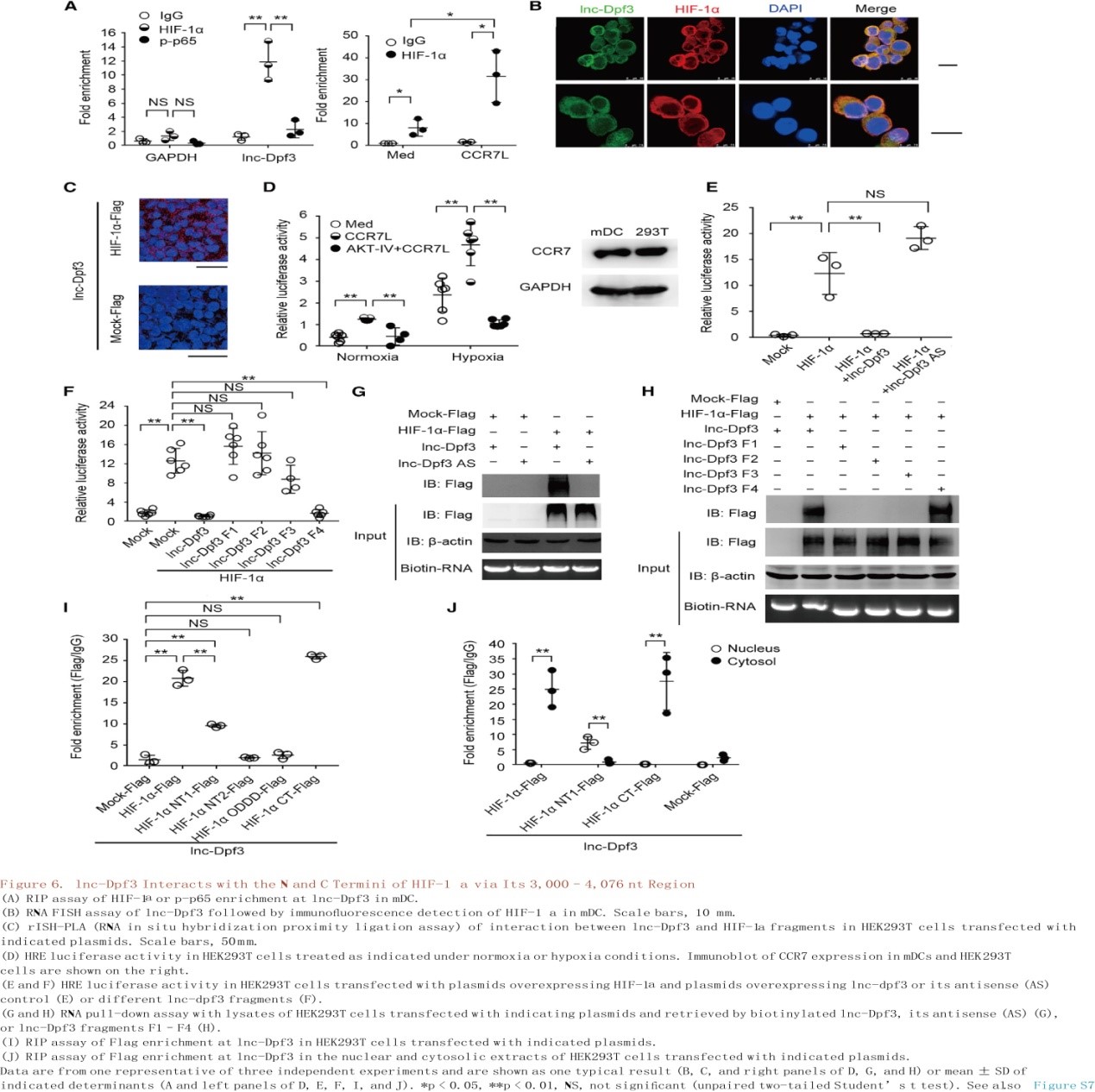
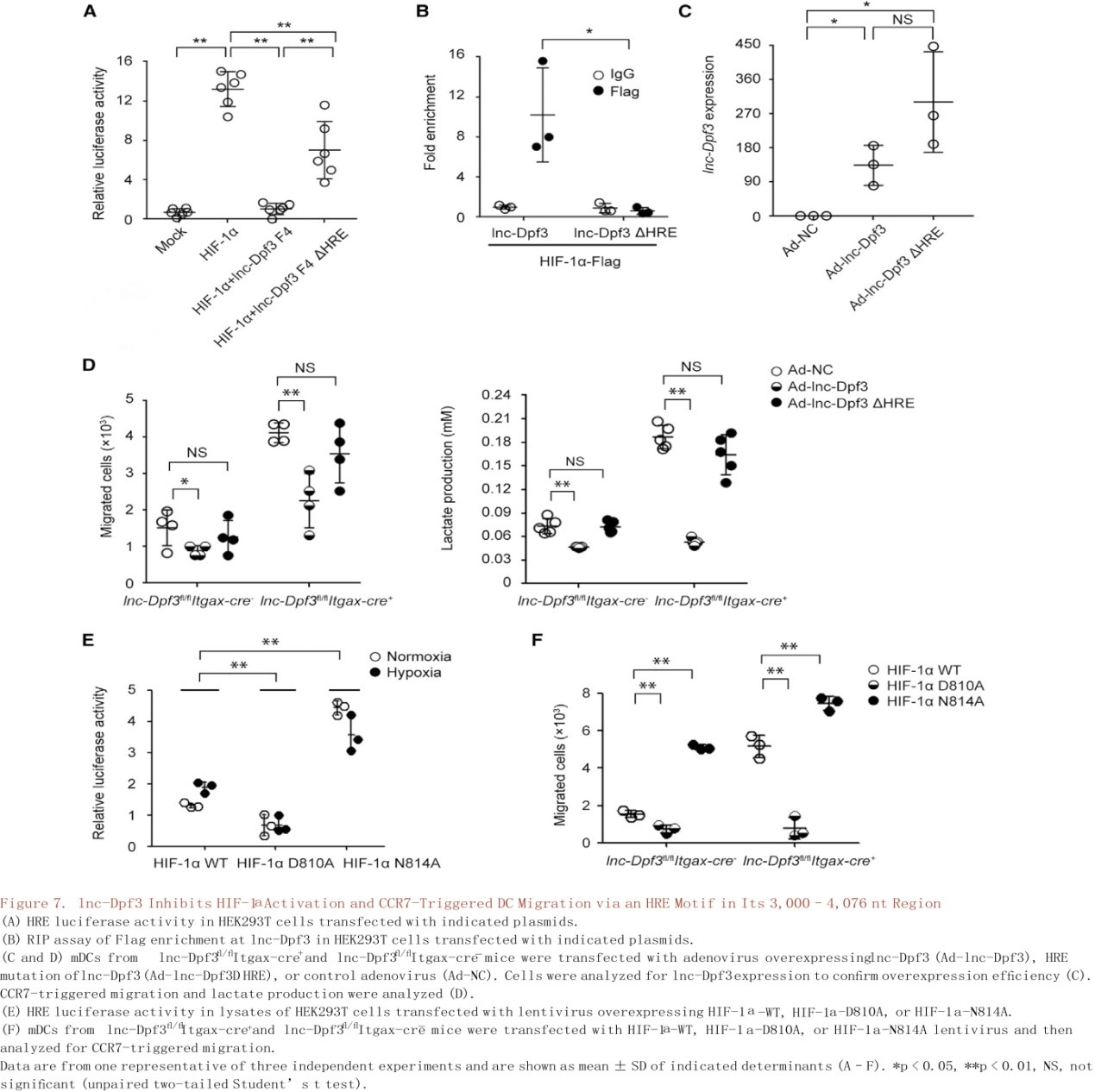
在确定lnc-Dpf3和HIF-1α之间的物理性连接后(图6A-C),使用缺氧反应元件(HRE)驱动的荧光素酶报告基因来测量HEK293T细胞中HIF-1α的活性(图6D)。在常氧和缺氧条件下,CCR7的刺激均以依赖Akt方式增强HIF-1α活性(图6D)。lnc-Dpf3的过表达抑制了HIF-1α的激活(图6E)。作者将lnc-Dpf3分成四个片段来映射HIF-1α结合区域,只有F4片段(3000-4076 nt)在抑制HIF-1α活性方面与全长lnc-Dpf3相似(图6F),lnc-Dpf3全长和F4片段均直接与HIF-1α结合(图6G和6H),可见HIF-1α是lnc-Dpf3 F4片段的结合伴侣。lnc-Dpf3在F4片段中含有一个HRE基序位于3,686- 3,690 nt。利用lnc-Dpf3的△HRE突变(ACGTG突变成AAAAG),发现HRE-基序突变部分地削弱了F4片段抑制HIF-1α活性(图7A)并且消除了lnc-Dpf3与HIF-1α的结合(图7B)。腺病毒介导的lnc-Dpf3过表达可抑制CCR7介导的DC迁移和乳酸的产生,恢复lnc-Dpf3fl/flItgax-cre+小鼠mDCs中过度的DC迁移和乳酸产生(图7C和图7D)。总之,lnc-Dpf3与HIF-1α的N和C端通过一个HRE 基序在其3,000- 4,076 nt区域相互作用,该基序对lnc-Dpf3抑制HIF-1α的活性和调节CCR7介导的DC迁移具有重要作用。
lnc-Dpf3通过阻止糖酵解NAD+的产生,抑制CCR7介导的DC迁移
LDHA可在有氧糖酵解的最后一步催化丙酮酸转化为乳酸,并与NADH氧化成NAD+有关。用草氨酸抑制LDHA可消除lnc-Dpf3沉默或缺乏引起的DC迁移增加的作用,表明lnc-Dpf3通过抑制LDHA表达调节DC迁移。补充NAD+,但不补充乳酸(LA),可以在草氨酸存在下恢复mDCs的迁移(图S7G和S7H)。这些数据表明大量的NAD+产生介导了lnc-Dpf3沉默或缺失DC中CCR7触发的迁移能力增强。在CCR7刺激的DC中,NAD+的过量产生可能参与调节白细胞迁移的关键导体–肌动蛋白细胞骨架。共焦分析表明,抑制糖酵解或LDHA可以防止CCR7刺激引起的F-肌动蛋白极化,而补充NAD+可以逆转草氨酸的作用(图S7I)。草氨酸对LDHA的抑制导致F-肌动蛋白聚合的显著降低,而NAD+的补充逆转了草氨酸的作用(图S7K)。因此,糖酵解NAD+通过维持F-肌动蛋白极化和聚合来支持DC迁移,lnc-Dpf3通过阻止糖酵解LDHA依赖的NAD+产生来抑制DC迁移。
小结
本研究鉴定了一种CCR7诱导的lncRNA(lnc-Dpf3),其通过直接结合和抑制HIF-1α活性来抑制糖酵解,从而抑制CCR7介导的DC迁移。在CCR7刺激后,lnc-Dpf3在后期的DC中明显上调。同时,lnc-Dpf3在CHS反应后期抑制DC迁移,并在1~3天抑制炎症损伤。lnc-Dpf3对抑制DC迁移晚期的延迟和诱导功能对于预防炎症发病和维持免疫平衡至关重要。新出现的证据表明,代谢重新编程在免疫细胞激活和调节中发挥着多重作用,但是人们对其潜在机制知之甚少。本研究通过代谢组学分析、生化研究和功能研究证实,CCR7刺激可通过激活HIF-1α诱导DC糖酵解反应,而HIF-1α激活受CCR7诱导的lnc-Dpf3抑制,从而预防炎症损伤。CCR7诱导m6A修饰下调,增加了lnc-Dpf3的表达,从而激活了lnc- Dpf3介导的DC迁移,防止了过度炎症反应的发生。因此,本研究为信号诱导的糖酵解反应如何反过来影响免疫细胞迁移和免疫反应提供了新的模型,并揭示了表观遗传机制和代谢途径在抑制炎症反应中的相互作用。
本研究非靶向代谢组学部分系由麦特绘谱承担检测,基于JiaLib自建数据库和全自动化XploreMet数据处理软件的质谱平台将持续助力广大医学科研人员,协助您们的研究工作迈向新途!
参考文献
Juan Liu, Xiaomin Zhang, Kun Chen, …, Xuetao Cao. (2019). CCR7 Chemokine Receptor-Inducible lnc-Dpf3 Restrains Dendritic Cell Migration by Inhibiting HIF-1a-Mediated Glycolysis. Immunity 50, 1–16.

