
文献解读
代谢流客户案例 | NSD1介导的PPARγ甲基化:撬动子宫内膜癌糖酵解重编程的关键“表观刹车”
子宫内膜癌(Endometrial Cancer, EC)作为最常见的妇科恶性肿瘤之一,其发病率持续攀升,背后隐藏着复杂的分子机制。代谢重编程是肿瘤细胞的显著特征,尤其是Warburg效应,使得肿瘤细胞在有氧条件下仍偏好糖酵解途径,以满足其快速增殖的能量需求。然而,驱动子宫内膜癌代谢异常的具体分子机制仍不明确,限制了其代谢治疗策略的开发。
NSD1,作为一种关键的组蛋白甲基转移酶,其在多种癌症中的突变和表观遗传沉默已引起广泛关注。尽管NSD1在子宫内膜癌中频繁突变,但其如何调控肿瘤代谢及肿瘤进展的具体机制尚不清楚。
2026年,同济大学附属上海市第一妇婴保健院研究团队在Cancer Research上发表了题为“NSD1-Mediated PPARγ Methylation Enhances PTEN Activity to Suppress Glycolysis and Tumor Progression in Endometrial Cancer”的研究,首次系统揭示了NSD1通过“甲基化-核定位-转录激活”轴驱动PPARγ/PTEN信号、抑制糖酵解和肿瘤进展的分子机制,提出了靶向NSD1酶活性或下游AKT的治疗新策略。(麦特绘谱提供代谢流([U-13C6]-glucose)技术检测服务)
研究思路
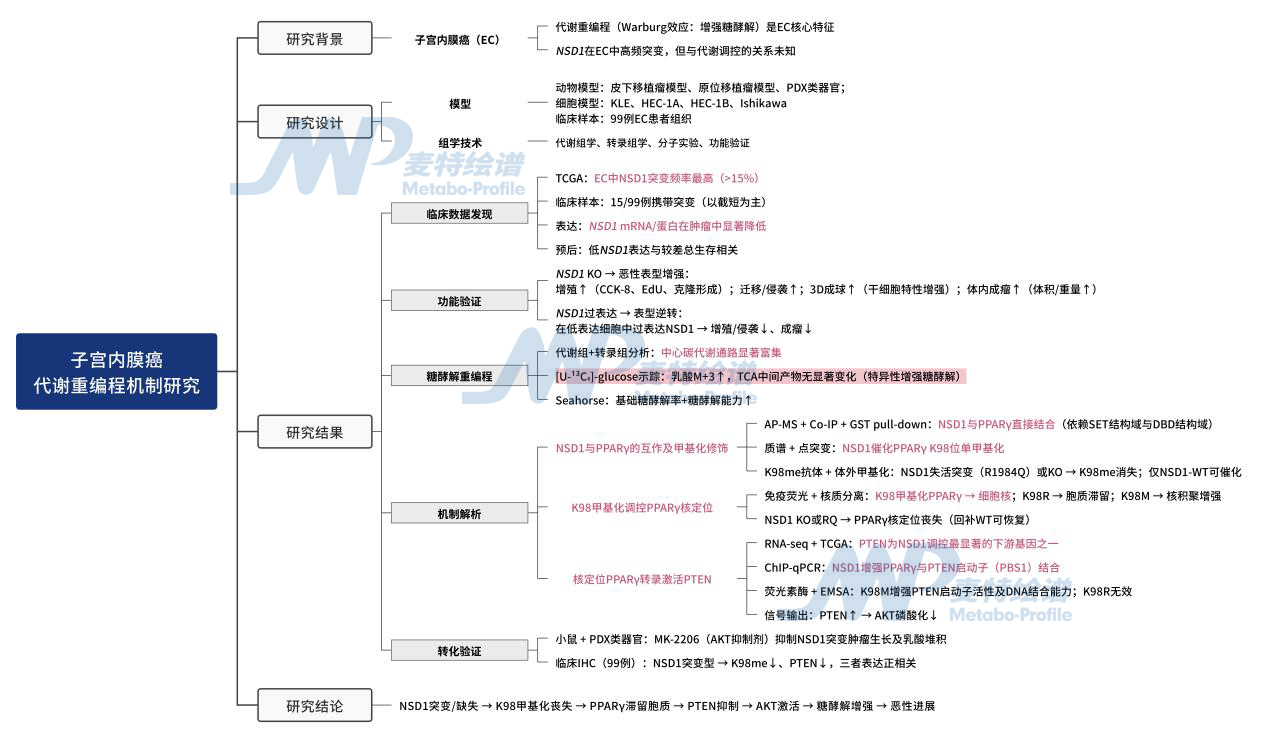
图1. 技术路线
研究结果
NSD1在子宫内膜癌中高频突变、表达降低,与不良预后密切相关
为明确NSD1在EC中的临床相关性,研究者首先利用TCGA和cBioPortal数据库分析,发现EC中NSD1突变频率超过15%,为所有肿瘤类型中最高,以截短突变和MSI/POLE亚型富集为特征。临床样本Sanger测序进一步确认15/99例携带NSD1突变(以截短为主)。NSD1 mRNA和蛋白在肿瘤组织中显著降低,且低表达与较差的总体生存显著相关。上述结果提示NSD1在EC中发挥潜在的抑癌功能。
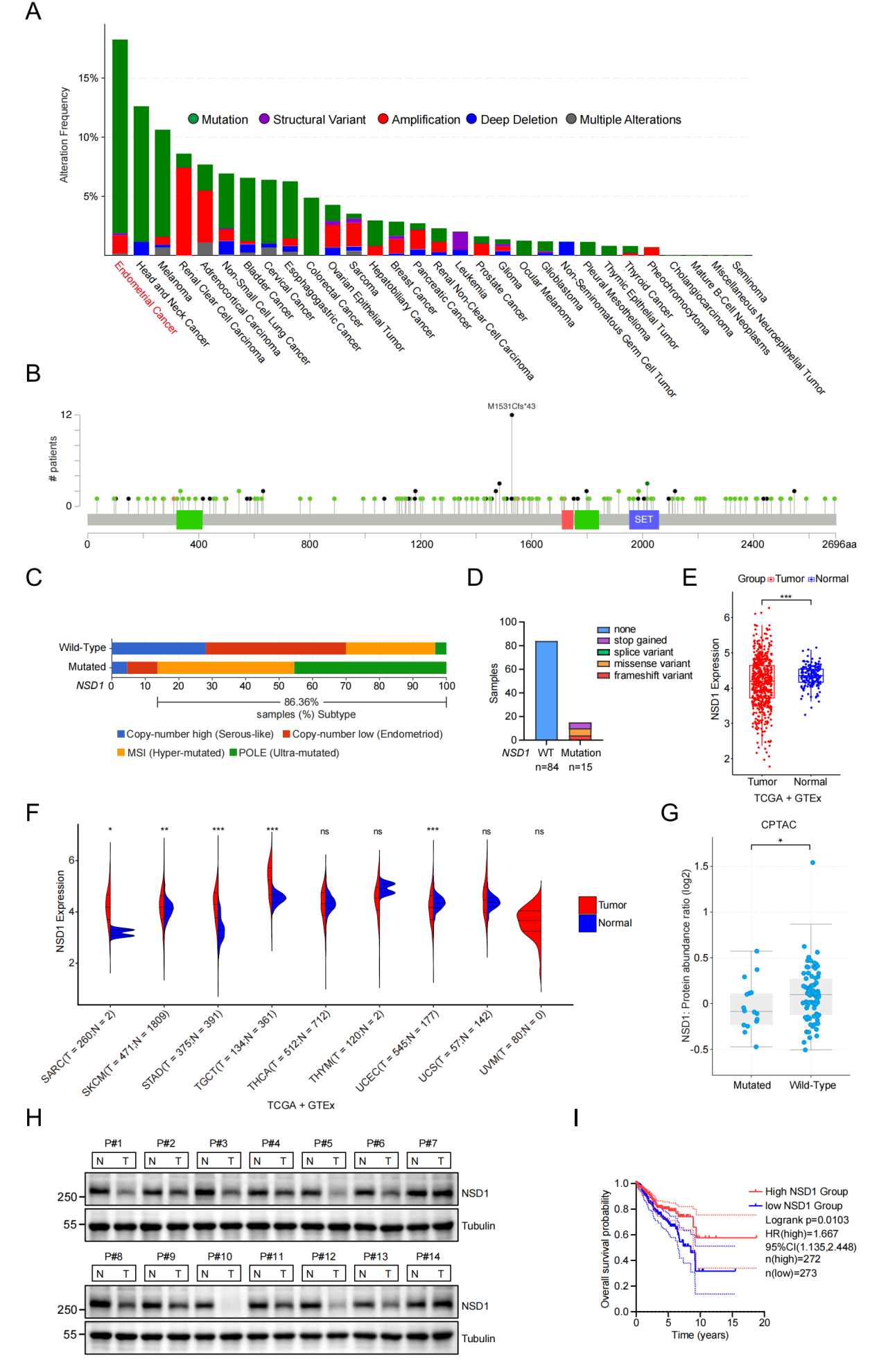
图2. NSD1在子宫内膜癌中高频突变并下调
NSD1缺失增强子宫内膜癌细胞增殖、侵袭与体内成瘤能力
利用CRISPR/Cas9构建NSD1敲除的KLE和HEC-1A细胞株,癌细胞出现明显恶性表型:增殖加快、克隆形成能力增强、EdU阳性率上升;迁移与侵袭能力显著升高;3D球体形成与体内成瘤速度加快、体积更大。反之,在NSD1低表达细胞(Ishikawa、HEC-1B)中过表达NSD1则可抑制上述恶性表型。证明NSD1是抑制子宫内膜癌恶性进展的核心分子。
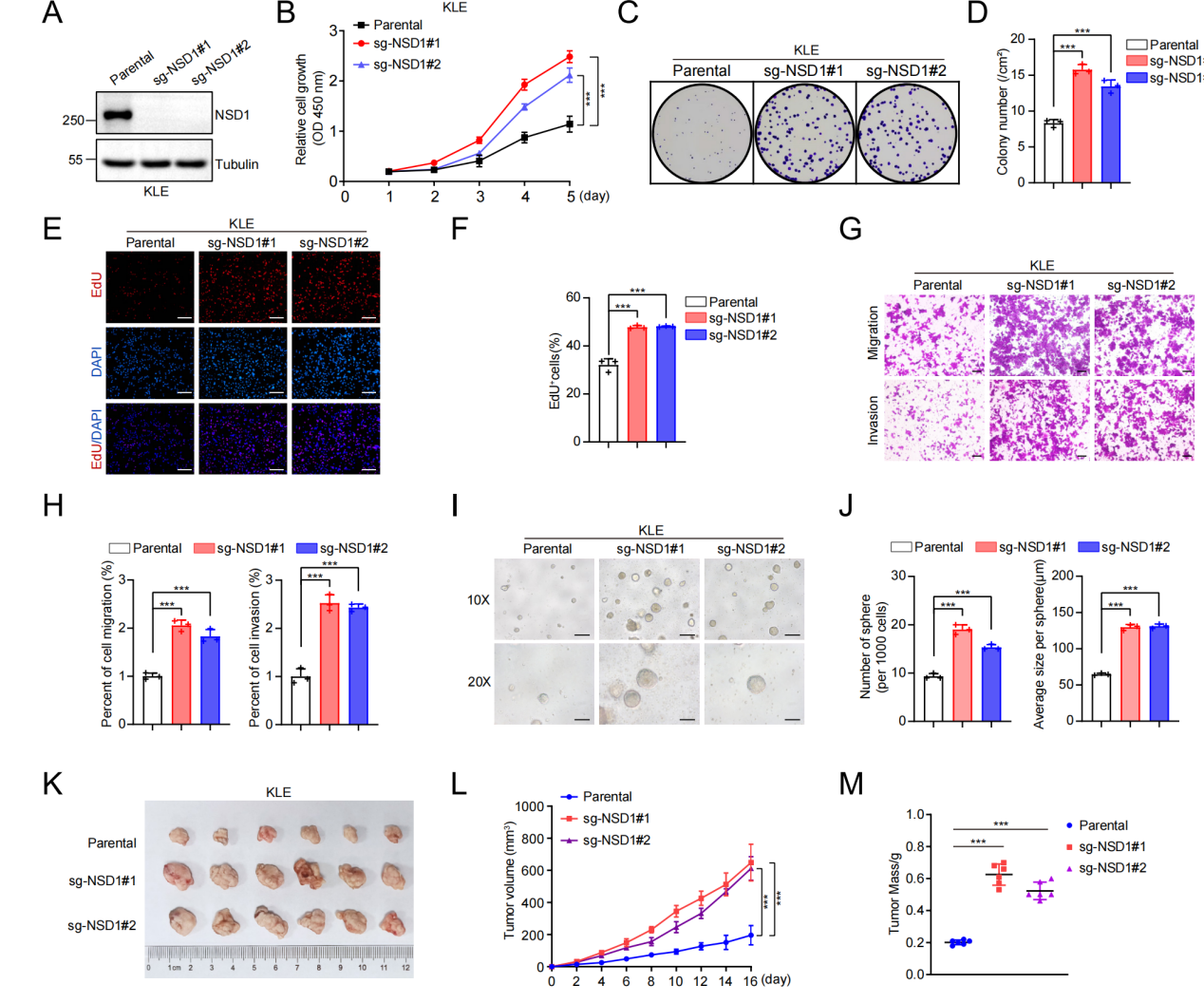
图3. NSD1缺失促进EC恶性表型
NSD1缺失驱动糖酵解重编程:代谢组+代谢流双验证
代谢组与转录组数据结果显示,NSD1敲除后中心碳代谢通路显著富集,且糖酵解相关代谢物明显升高。进一步通过麦特绘谱提供的[U-13C6]-葡萄糖同位素示踪代谢流分析,发现NSD1缺失导致乳酸M+3水平显著上升,而TCA循环中间产物(M+2柠檬酸等)无明显变化,提示糖酵解通量特异性增强;ECAR、葡萄糖摄取、乳酸分泌、胞内ATP均显著升高。反之,过表达NSD1则可抑制糖酵解。上述结果进一步明确NSD1是维持EC代谢稳态、抑制Warburg效应的关键分子。
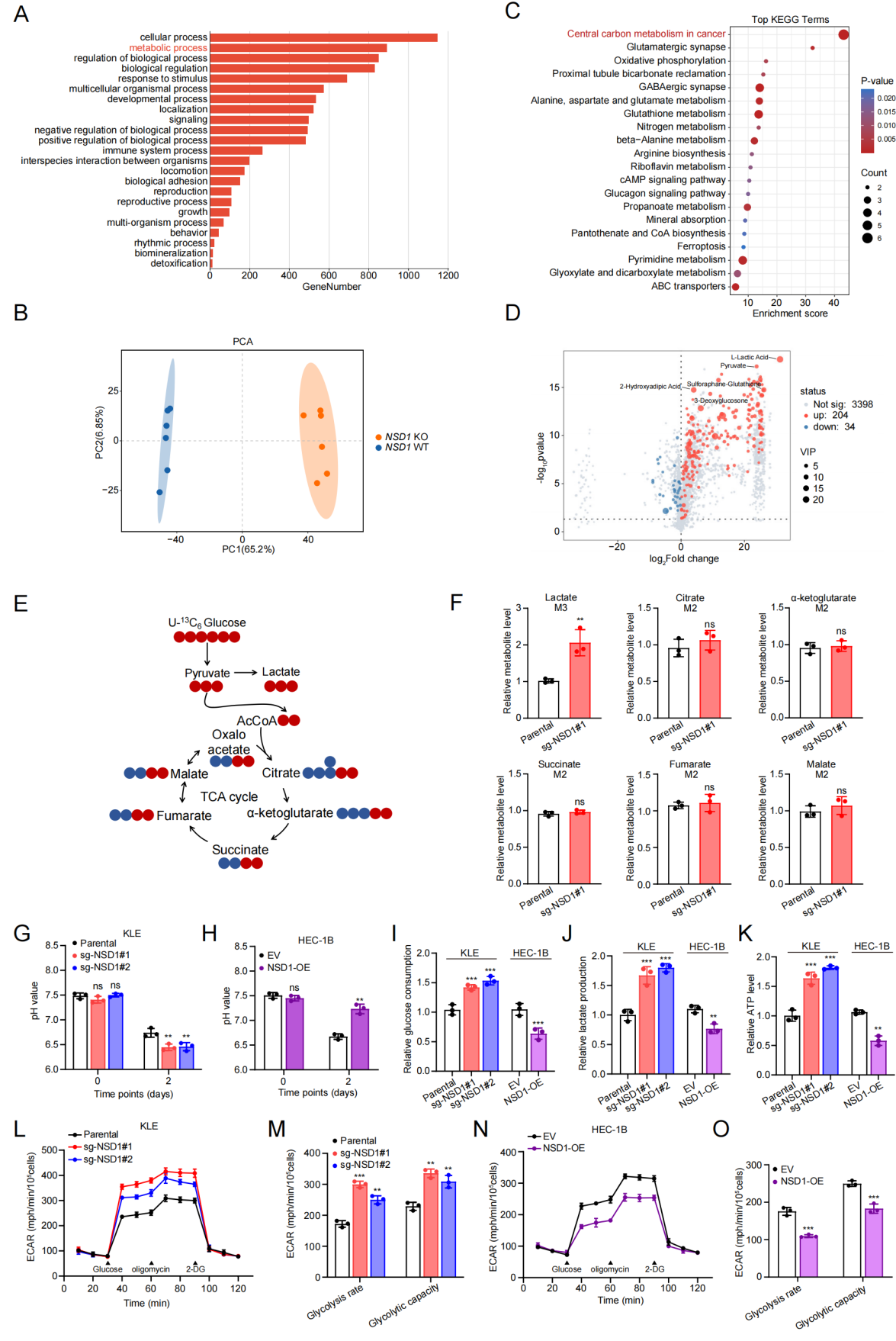
图4. NSD1缺失驱动癌细胞发生糖酵解重编程
NSD1直接结合PPARγ并催化其K98位点单甲基化
为寻找NSD1的非组蛋白底物,研究者进行了AP-MS及Co-IP,首次鉴定PPARγ为NSD1的直接互作蛋白,且结合依赖于NSD1的SET结构域和PPARγ的DBD结构域。GST pull-down确认两者直接结合。质谱分析及点突变验证显示,NSD1主要催化PPARγ第98位赖氨酸(K98)的单甲基化。自制K98me特异性抗体进一步确认:NSD1失活突变(R1984Q)或NSD1 KO可完全消除K98甲基化;而PPARγ激动剂罗格列酮可增强该修饰。体外甲基化实验证实,仅野生型NSD1(而非R1984Q)能催化PPARγ甲基化,确立了NSD1作为K98甲基化直接催化酶的地位。
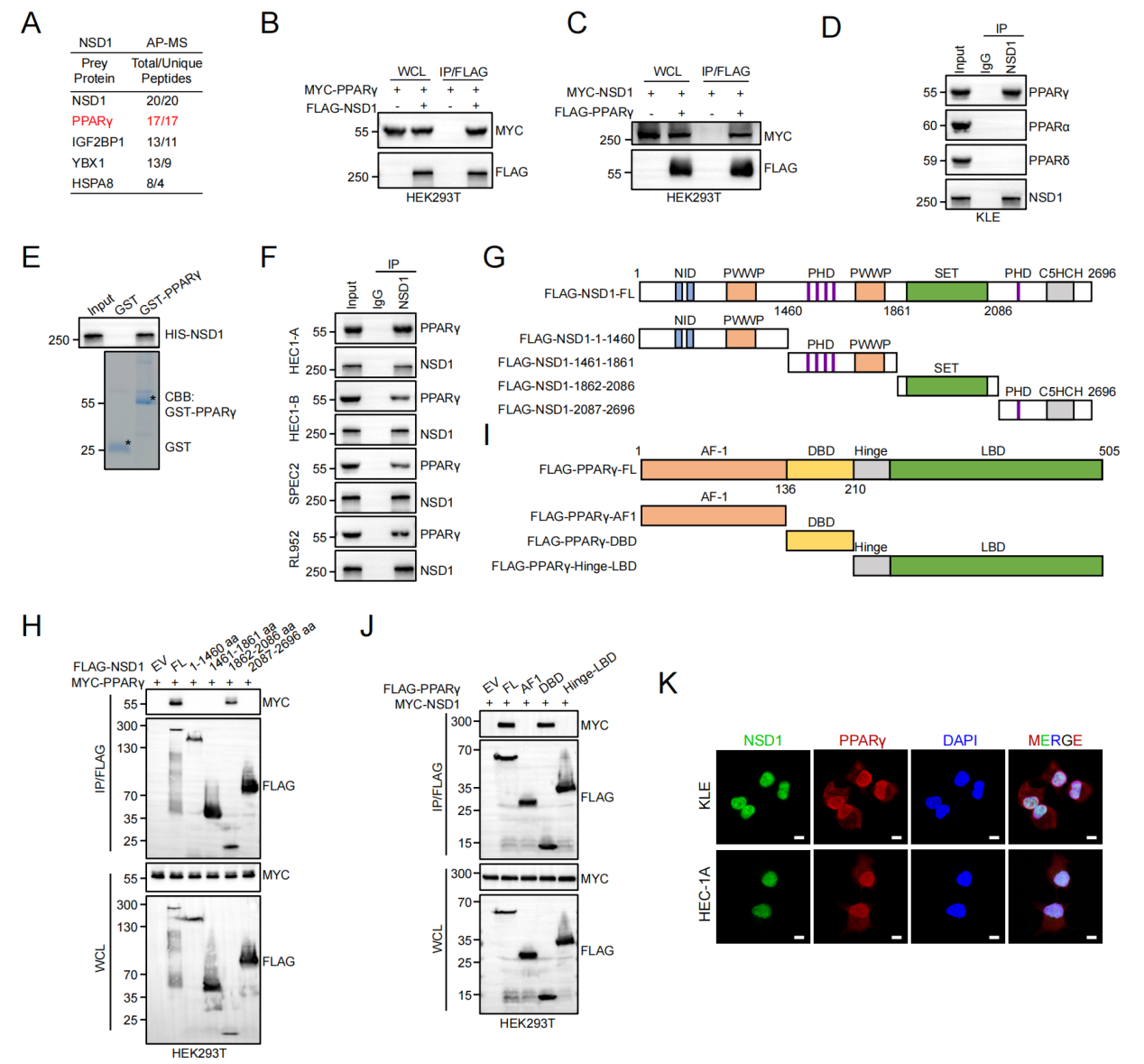
图5. NSD1直接结合PPARγ并催化其K98位点单甲基化
K98甲基化是PPARγ核定位与转录活性的“分子开关”
免疫荧光及核质分离实验显示,K98甲基化的PPARγ主要定位于细胞核;非甲基化模拟突变K98R导致PPARγ滞留于细胞质,而甲基化模拟突变K98M则显著增强其核积聚。NSD1 KO或催化失活突变体均导致PPARγ核定位丧失,而回补野生型NSD1可恢复。该发现首次揭示:K98甲基化是PPARγ核转位的关键“分子开关”。
在转录调控层面,RNA-seq和TCGA分析发现PTEN是NSD1调控最显著的下游基因之一,且NSD1与PTEN表达呈正相关。ChIP-qPCR和荧光素酶报告实验证实:NSD1催化PPARγ K98甲基化后,增强PPARγ与PTEN启动子(尤其PBS1位点)的结合,促进PTEN转录。K98M突变增强PTEN启动子活性及DNA结合能力(EMSA证实),而K98R突变则无效。PTEN上调后AKT磷酸化被抑制,构成了完整的“甲基化→核定位→转录→信号抑制”级联。
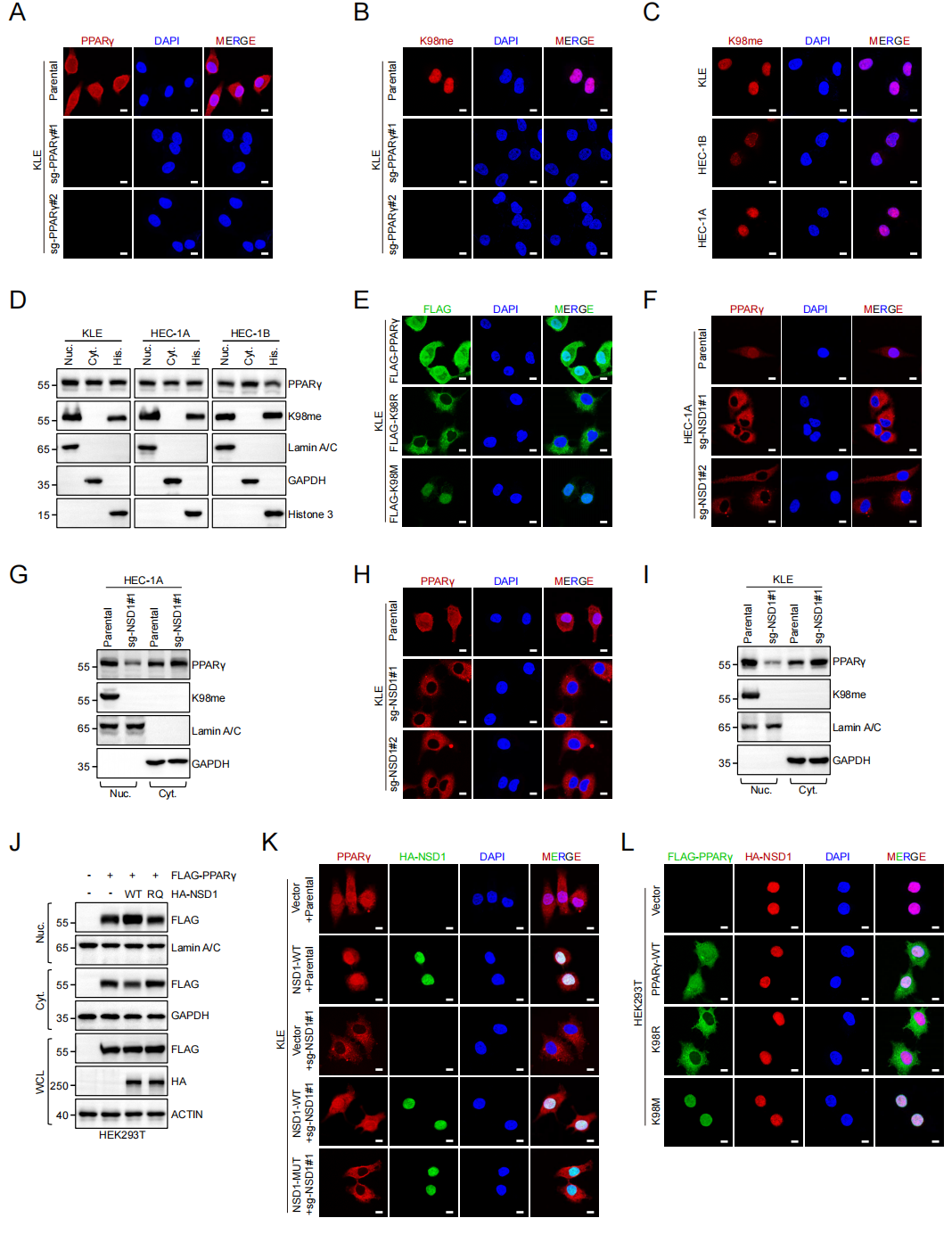
图6. K98甲基化决定PPARγ核定位与转录活性
NSD1–PPARγ轴通过转录激活PTEN抑制 AKT与糖酵解
功能必要性验证方面,使用NSD1特异性抑制剂BT5或回补催化失活突变体(NSD1-RQ)均无法抑制糖酵解、增殖及侵袭,而野生型NSD1可完全逆转NSD1 KO表型。这表明:NSD1的甲基转移酶活性是其抑癌功能的前提。
下游可靶性验证方面,在NSD1 KO细胞中过表达PTEN或使用AKT抑制剂MK-2206,可显著降低葡萄糖摄取、乳酸生成及ECAR水平,并抑制细胞增殖和侵袭能力。在小鼠移植瘤模型及PDX类器官中,MK-2206同样可抑制NSD1突变肿瘤的生长和乳酸堆积。临床IHC分析显示:NSD1突变型EC患者肿瘤组织中K98甲基化PPARγ及PTEN表达显著降低,且三者表达呈正相关。这些数据共同证明:PTEN下调及AKT过度激活是NSD1缺失导致代谢重编程和肿瘤进展的核心下游事件,且该轴具有明确的治疗可靶性。
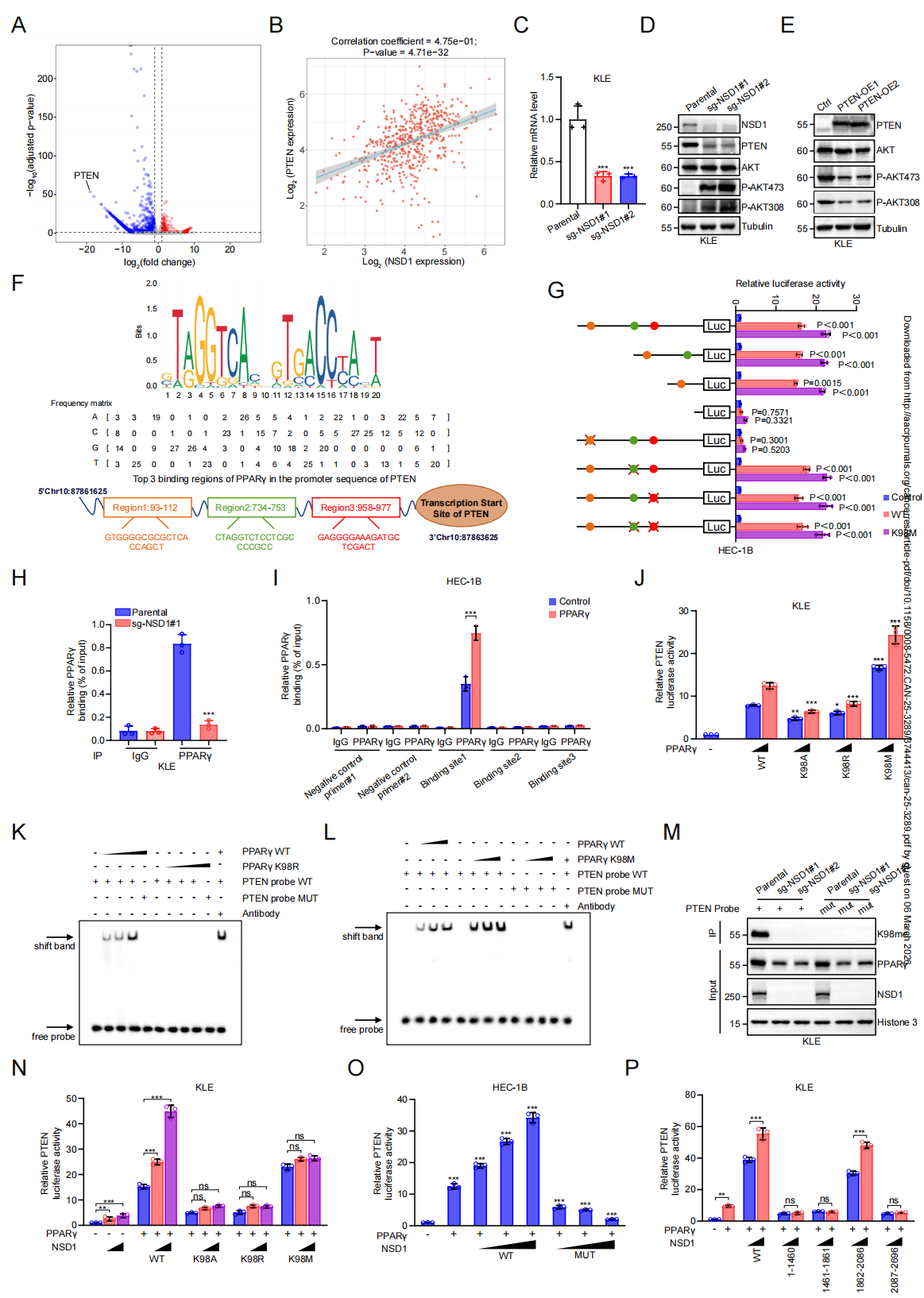
图7. NSD1通过PPARγ甲基化促进PTEN转录,抑制AKT信号
研究结论
本研究首次完整揭示子宫内膜癌中 “表观–代谢–肿瘤进展” 的全新调控轴:NSD1缺失→PPARγ K98甲基化丧失→PPARγ胞质滞留→PTEN转录抑制→AKT过度激活→糖酵解增强→肿瘤增殖侵袭增强→预后不良,将NSD1–PPARγ–PTEN定位为子宫内膜癌代谢表观核心开关,提出AKT抑制剂、PPARγ激动剂、代谢联合治疗的全新策略,为子宫内膜癌的靶向与精准治疗提供了突破性机制与转化方向。
参考文献
NSD1-Mediated PPARγ Methylation Enhances PTEN Activity to Suppress Glycolysis and Tumor Progression in Endometrial Cancer. Cancer Research. 2026
请扫描二维码阅读原文

绘谱帮你测
麦特绘谱为本研究提供的 [U-13C6]-glucose 代谢流分析显示,NSD1缺失可显著升高乳酸M+3水平,而TCA循环中间产物(M+2柠檬酸等)无明显变化,提示糖酵解通量特异性增强。这一代谢特征结果为NSD1缺失通过驱动糖酵解重编程,促进子宫内膜癌细胞的恶性增殖与侵袭提供了关键依据。
麦特绘谱开创性地搭建了医学领域高端代谢组学技术平台,覆盖了非靶向-全定量-代谢流等全方位的高端医学代谢组解决方案,同时全面布局微生物组学、转录组学和蛋白质组学等多组学技术服务,已成为全球多组学研究者的优选合作伙伴。麦特绘谱已为数百家三甲医院、科研院所和企业提供多组学一站式整体解决方案,协助客户与合作伙伴发表SCI文章600+篇,累计影响因子6000+,平均IF>10,涵盖Cell, Science, Nature, Cancer Cell, Signal Trans-duction and Targeted Therapy, Nature Biotechnology, Cell Metabolism等顶级期刊。
