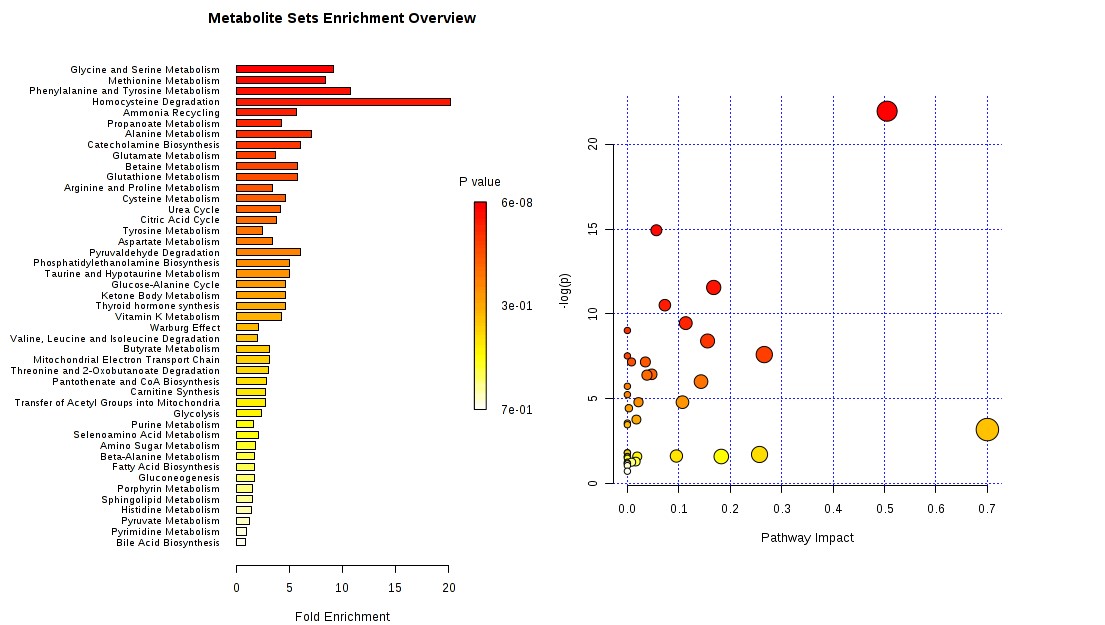
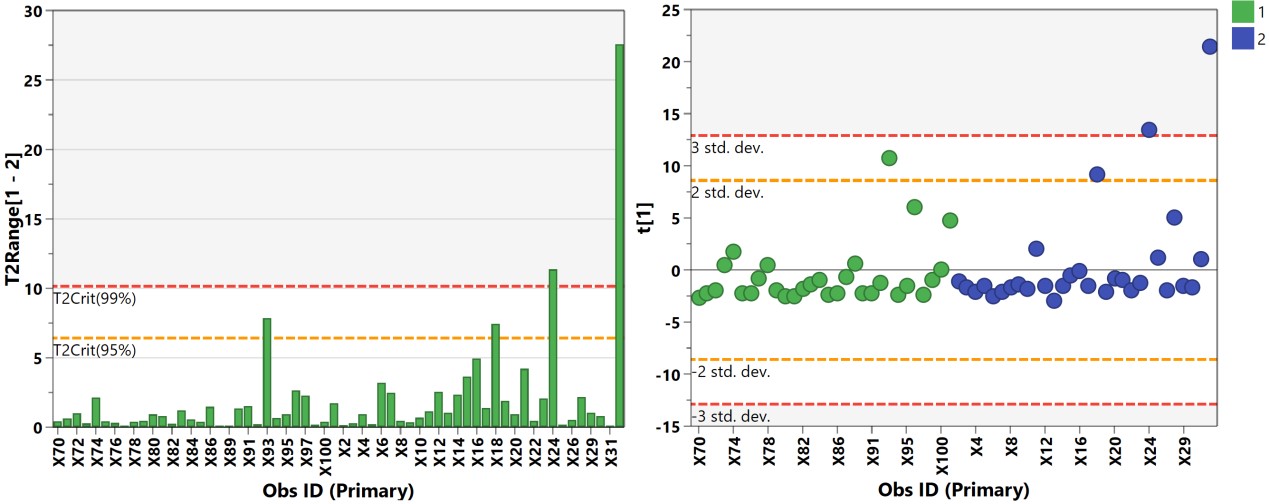
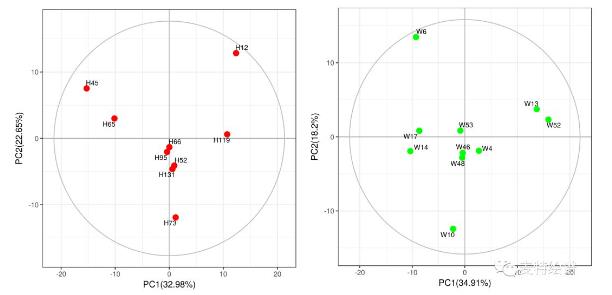
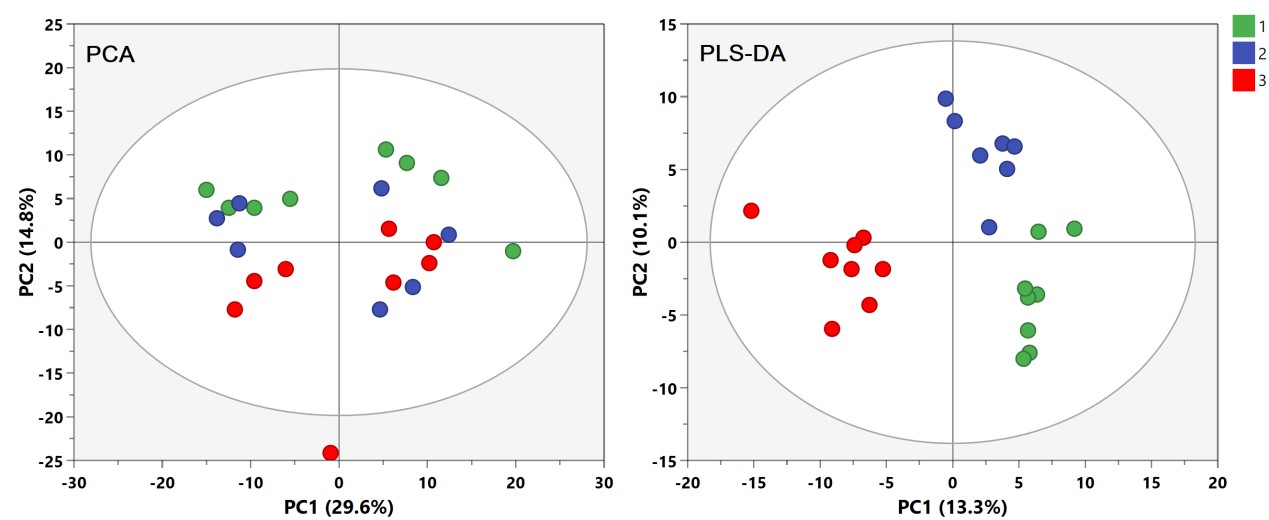
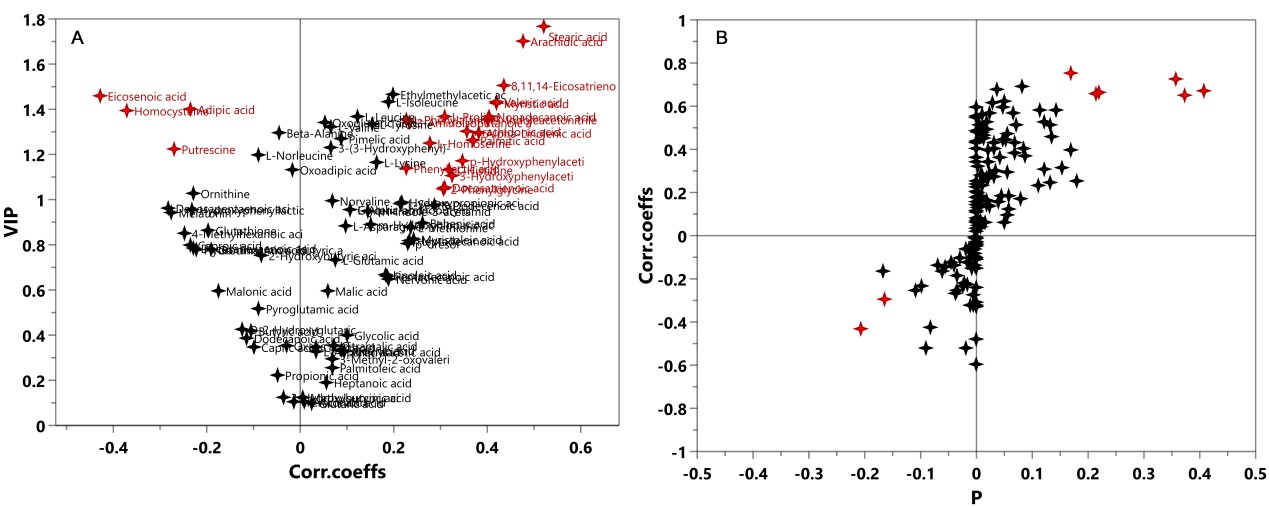
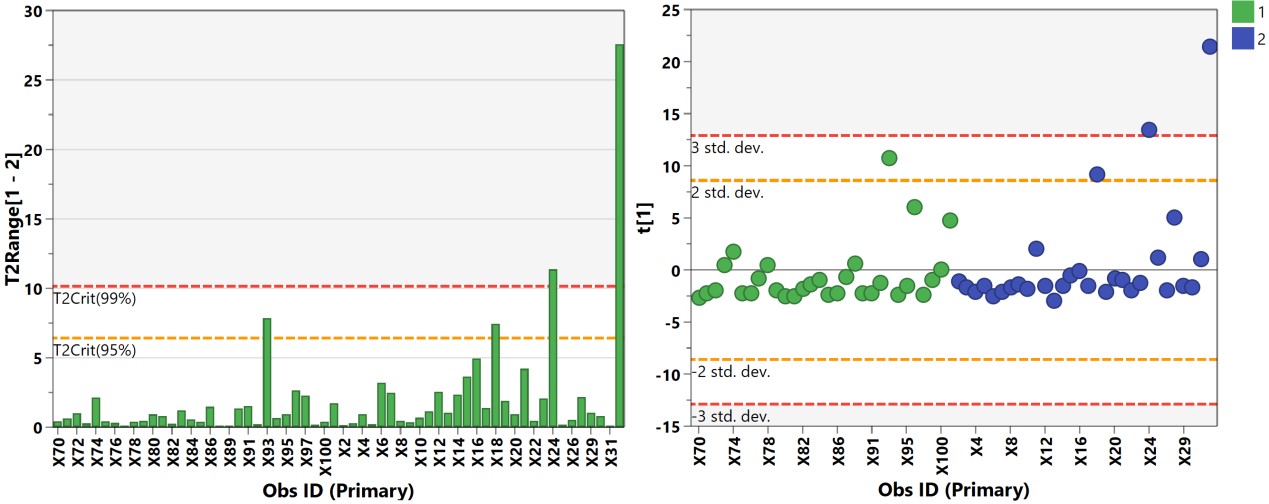
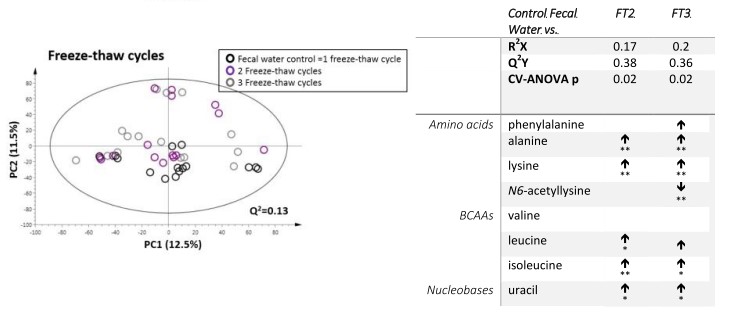
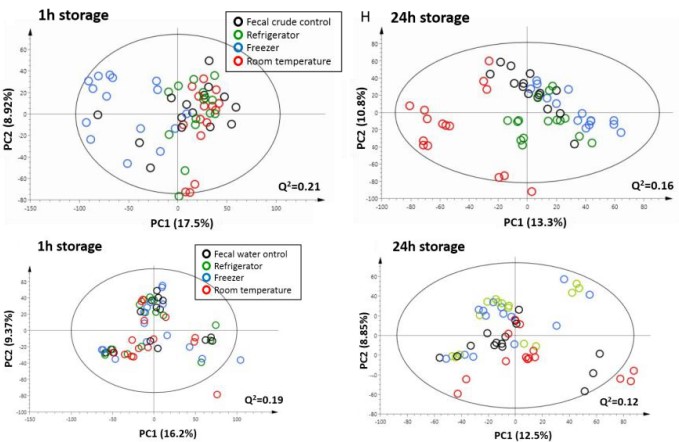
有监督的分类模型缺点是可能会出现过拟合(over-fitting)现象,即模型可以很好地将样本进行区分,但用来预测新的样本集时却表现很差。因此对于有监督的分类模型,我们需要验证模型的可靠性,下面列出几种常见的模型评价方法:
1. K折交互验证(K-fold cross validation)
最可靠的方式是将数据分为训练集(Training set)、验证集(Validation set)和测试集(Test set),训练集用于训练模型,验证集优化模型,测试集测试模型的预测能力。但受限于样本数量,通常采用K折交互验证。其中七折交互验证较为常用,即将数据集分为7份,每次挑选出1份作为测试样本,剩余的6份用来训练建模,整个过程将会被重复直到所有样品都被预测过。预测的数据将会和原始数据作对比得到预测残差平方和(Predicted residual sum of squares, PRESS)。为方便起见,将PRESS转变为Q2(1-PRESS/SS)。Q2越大表示模型的预测能力越好。对于生物学样本,Q2≥0.4是比较理想的[2],Q2≥0.2往往也可以接受,只是模型比较弱。软件在自动建模(Autofit)时,会根据Q2决定模型所用的主成分或Orthogonal component个数(OPLS-DA模型)。当Q2停止增长时,模型将不再增加主成分。
2. 置换检验(Permutation test)
仅用Q2仍不足以证明模型的可靠性,置换检验也是常用的模型评判方式,常和Q2结合使用。其原理是将每个样本的分组标记随机打乱,再来建模和预测。一个可靠模型的Q2应当显著大于将数据随机打乱建模后得到的Q2。基于置换检验的结果,可以画出Permutation plot(图6)。该图展示了置换检验得到的分组变量和原始分组变量的相关性以及对应的Q2值,虚线为回归线。一个可靠的有监督模型要求回归线在Y轴上的截距小于0。
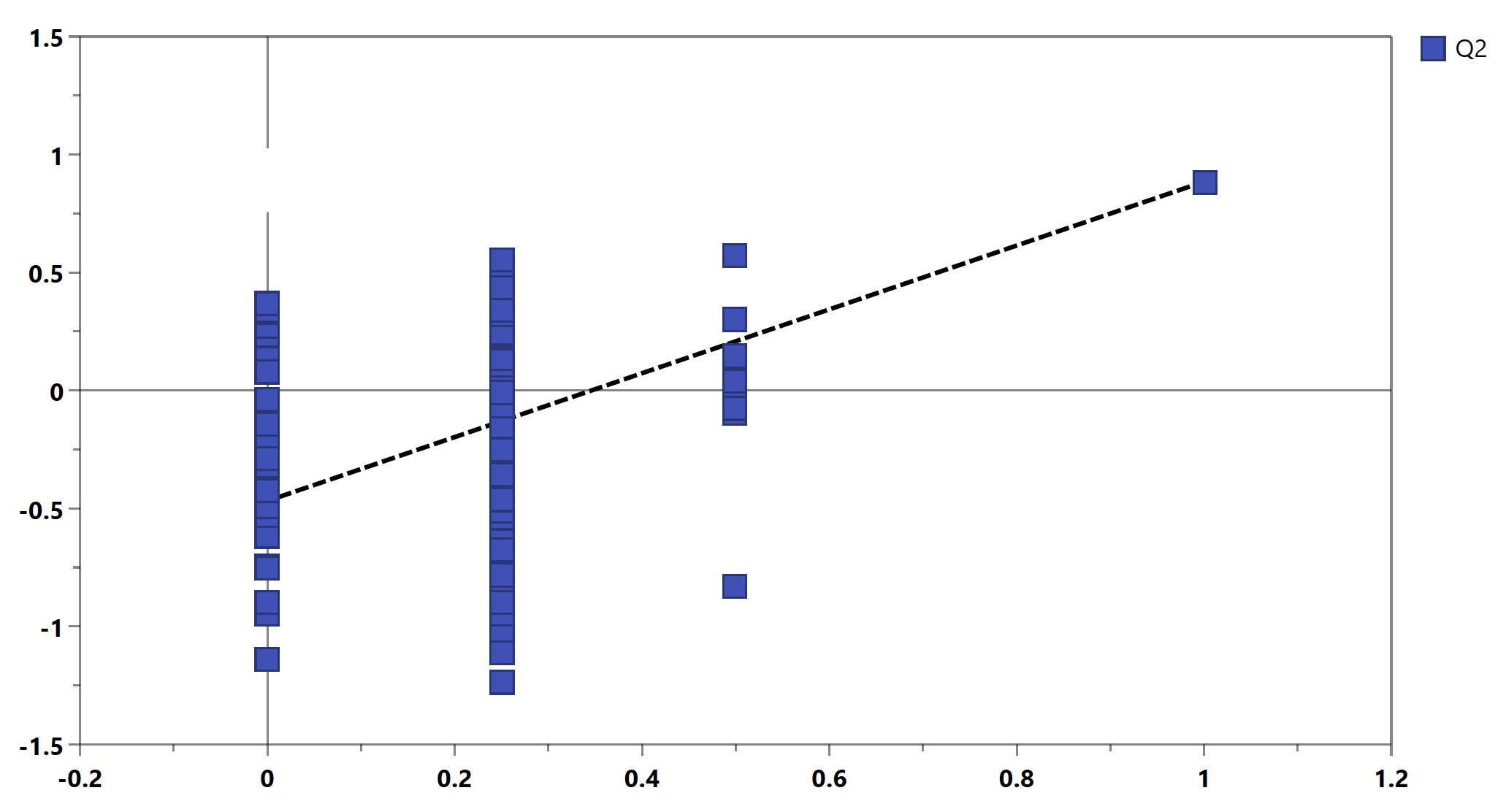
图6. Permutation plot用于模型验证
3. 基于交互验证的方差分析(CV-ANOVA)
CV-ANOVA是基于交互验证预测残差的方差分析,利用方差分析测试预测的Y变量(Yhat)和预设Y变量(Yobs)的残差和Yobs围绕均值变化的差异。它的好处是可以将交互验证的结果以更加熟悉的方式展现出来,输出表征统计学意义的P值。但CV-ANOVA对于小样本集的检验效能较低[3]。